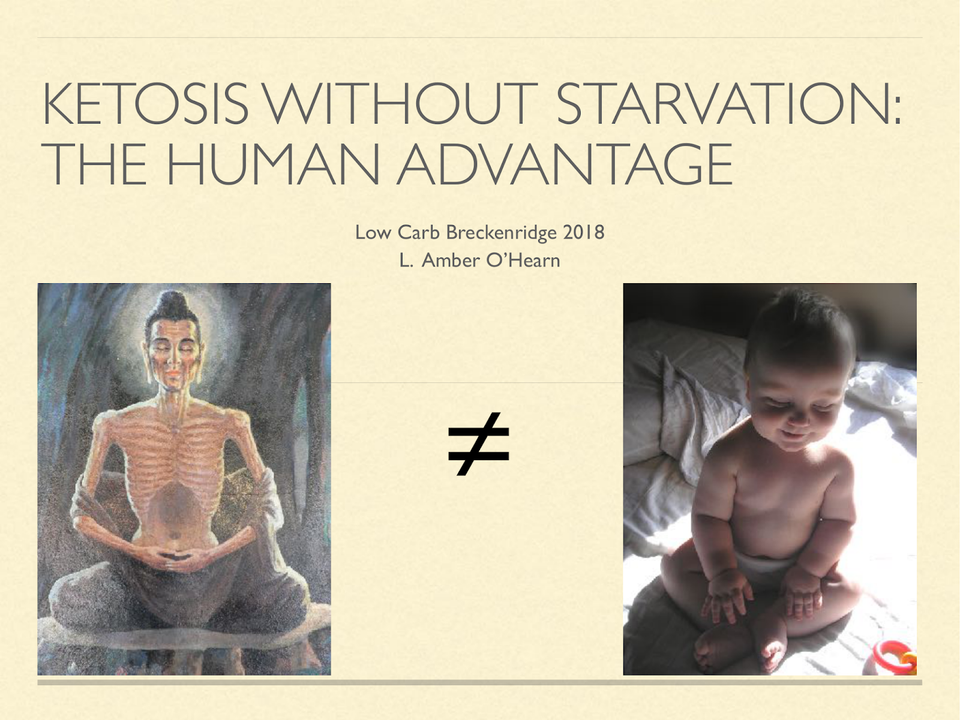
Ketosis Without Starvation: the human advantage
I recently had the honour to speak at Low Carb Breckenridge 2018.
You can find the video here. Below are my slides, notes, and references.
On a high carb diet, you might need to fast to attain an enlightened brain state.On a ketogenic diet, as a human, that doesn’t appear to be necessary.
The only disclosure I have to declare is that I have some generous supporters on Patreon for my writing. Thank you!
The supported content is free, so these are donations.

The foundation of our biochemical understanding of ketosis came from experiments in fasted humans and other animals.
For example the groundbreaking work of George Cahill.
I recommend his publication Fuel Metabolism in Starvation ([Cah2006]) which reviews many of his findings.
We continue to learn about mechanisms for how ketosis may increase health in a variety of ways.
However, these origins carry with them an implicit cautionary note, since starvation is generally not recommended, for obvious reasons.
It’s not sustainable indefinitely. It’s stressful to the body. And it can do real harm, sometimes with lasting detrimental consequences.
Even fasting for short periods is surrounded by controversy among experts at this very conference, because of its potential to do damage to lean mass, and all the potential problems of protein and calorie malnutrition. If ketosis is like fasting, we had better use it carefully, judiciously, and sparingly.

Many researchers conceptualise metabolism as operating in two complementary phases.
The act of eating or not eating sets off a cascade of hormonal and molecular signals that result in one phase or the other, sometimes called the fed and fasting states. In this paper [Mat2018] they are called the glucose and ketone phases.
Important things happen in both phases.
The fed state is attributed with generating and synthesising things like tissue, mitochondria, and neurons, but the fasted state is attributed with clearing broken structures for renewal and repair, and providing the stimuli to direct the synthesis phase. Ketosis is normally indicative of the fasting state.
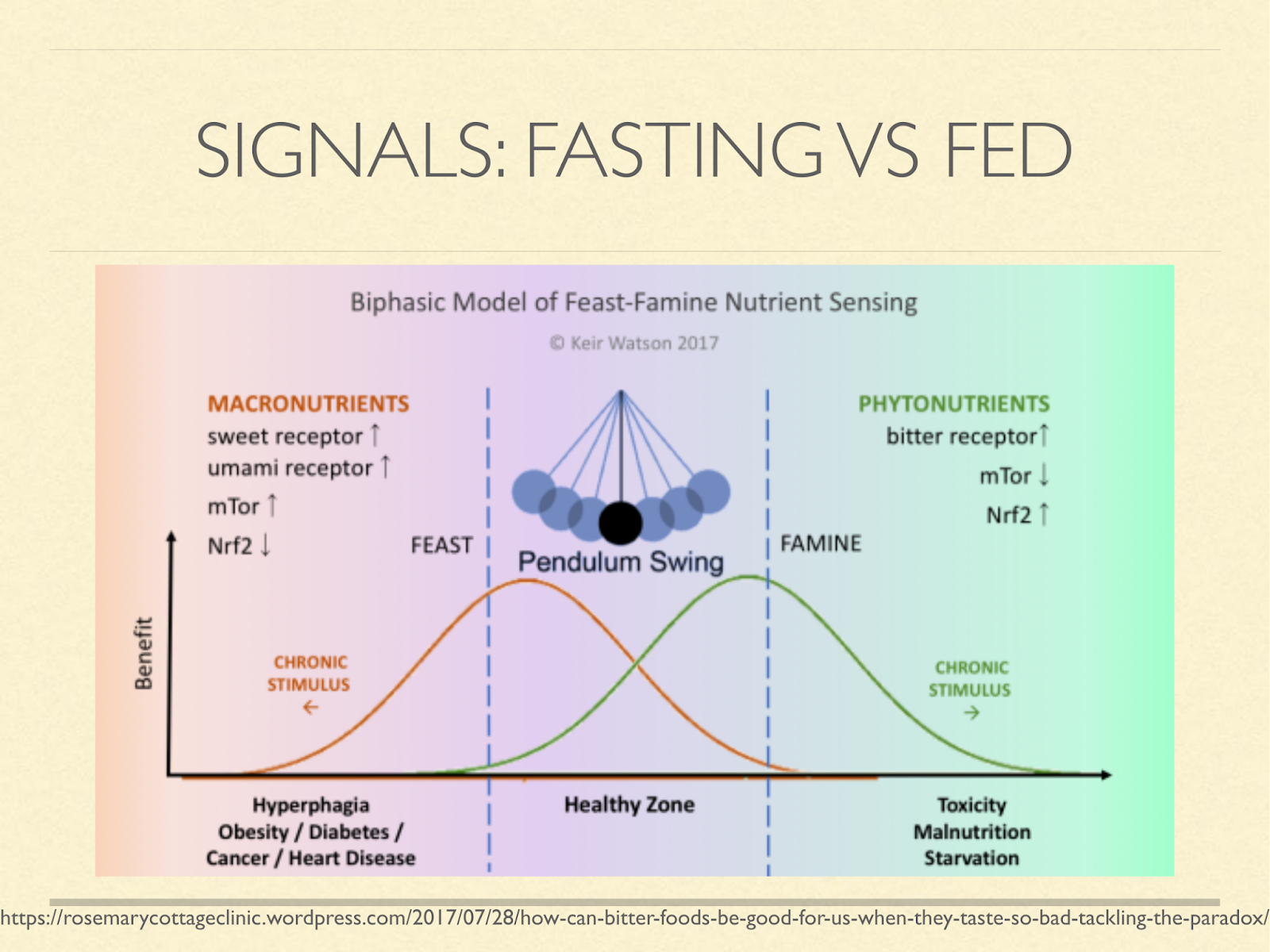
Many believe that staying in either phase prolongedly leads to disease.
And so you will hear people talk about metabolic switching, metabolic flexibility, insulin pulsatility, and so on.
Ketosis is normally an indication and a signal of the fasting state, so reason tells us that chronic long-term ketosis is unhealthy.
[Graphic from a blog post on the role of bitter in enhancing the starvation signal. https://rosemarycottageclinic.wordpress.com/2017/07/28/how-can-bitter-foods-be-good-for-us-when-they-taste-so-bad-tackling-the-paradox/]
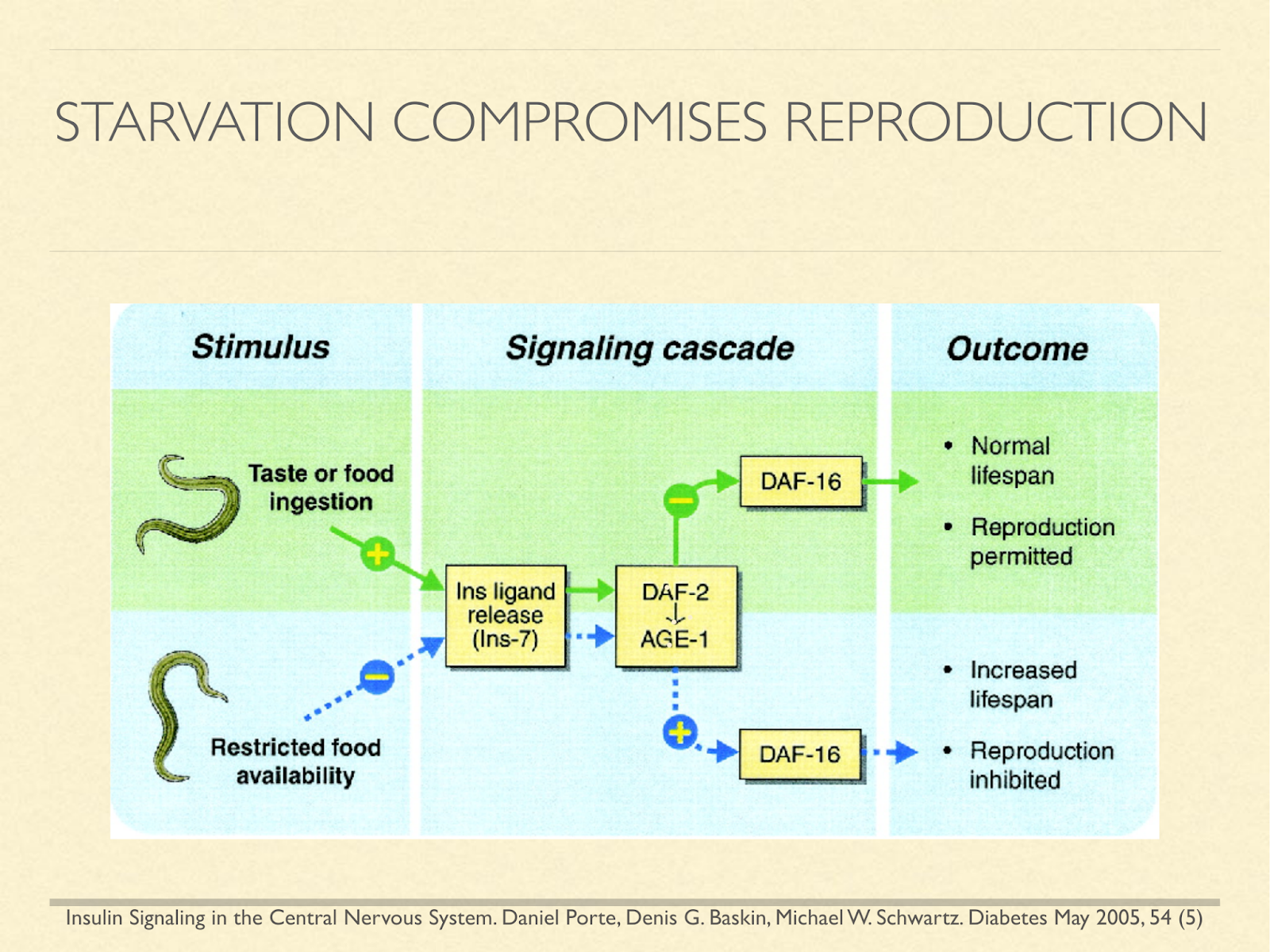
Further, it’s been shown in longevity research that many animals use signals of fed and fasting state to determine whether to reproduce, because it’s a time of plenty, or to slow aging and shut down reproductive ability until more favorable conditions arise. So again, the comparison leads us to fear that ketosis may have benefits, but that it comes with a severe cost.
[Graphic from: Insulin Signaling in the Central Nervous System. Daniel Porte, Denis G. Baskin, Michael W. Schwartz. Diabetes May 2005, 54 (5)]

But hold on. It turns out that starvation is not the only condition where ketosis naturally arises. Fetuses use ketones in the womb [Sha1985], [Ada1975], [Cun2016].
The placenta is full of BOHB [Mun2016]. Some mammals, humans included, have “ketosis of suckling”. Breastfed infants are in mild ketosis [Per1966], [Kra1974], [Bou1986].
In fact humans of all ages easily attain ketosis without protein or calorie deprivation, so long as they aren’t eating carbohydrates.
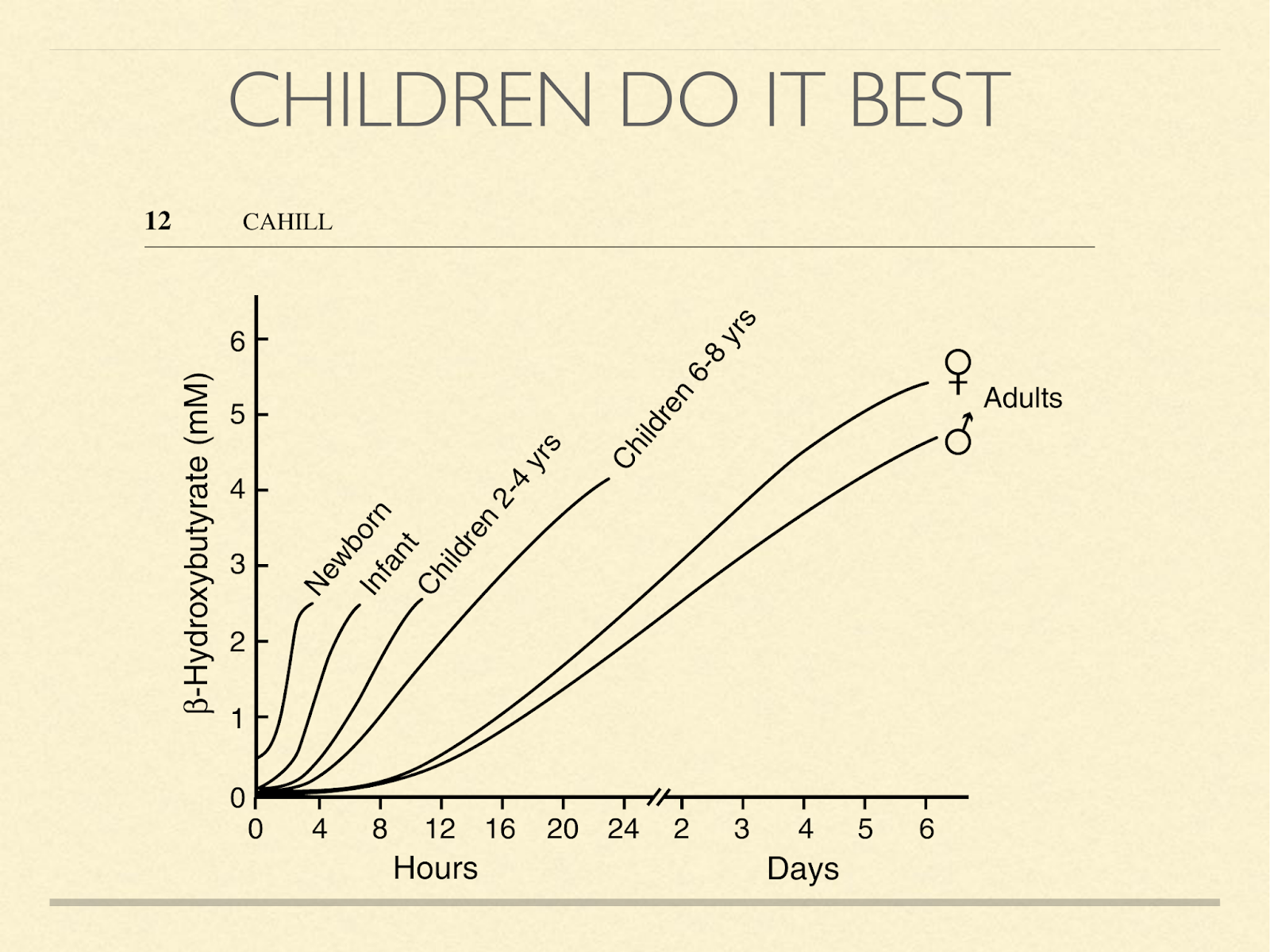
This graph shows how quickly the concentration of BOBH goes up in humans when they stop eating. It’s inversely related to age, one of the stunning things about it is the orders of magnitude involved. Look for example at the 6-8 year old children. If the 4-hour mark is about 0.1 – 0.2, then in a day, it’s increased by a factor of 20 or 40. Newborns, who typically aren’t yet eating cereal, of course, don’t start that low. Also notice that children don’t even need to miss a day of food to get above the 0.5 mmol level of ketosis which has been considered the threshold of nutritional ketosis by Phinney and others. In his presentation here, he has even said that benefits likely begin even below that level.
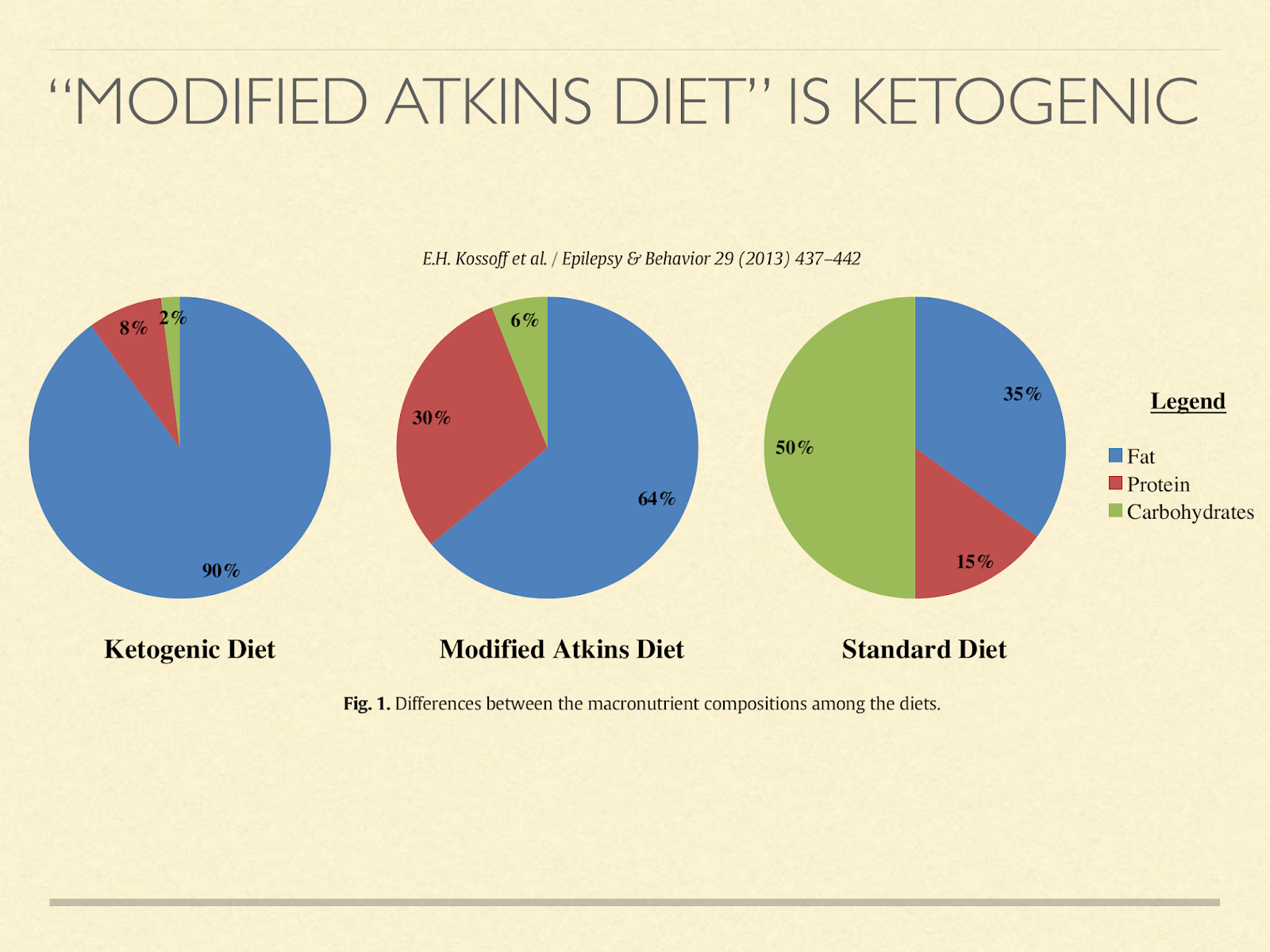
But they don’t have to abstain from eating for ketosis to happen. For example, we have results in epileptic children. The previous standard had been a tightly protein restricted ketogenic diet. We now know that most children don’t need that for seizure control. Eating a modified Atkins diet, which mostly just means they stay in the induction phase instead of adding back carbs, typically they are in ketosis, even though they eat ad libitum [Kos2013]. These are growing children and adolescents.
Unlike with the protein restricted versions of ketogenic diets for epilepsy, which in some cases have impacted growth, when protein isn’t restricted, neither is growth [Nat2014].

Even adults have this ability. To know whether adults are able to stay in ketosis when protein needs are exceeded,
we have to know what our protein needs are. It depends who you ask.
[Please see also How much protein is enough? ]
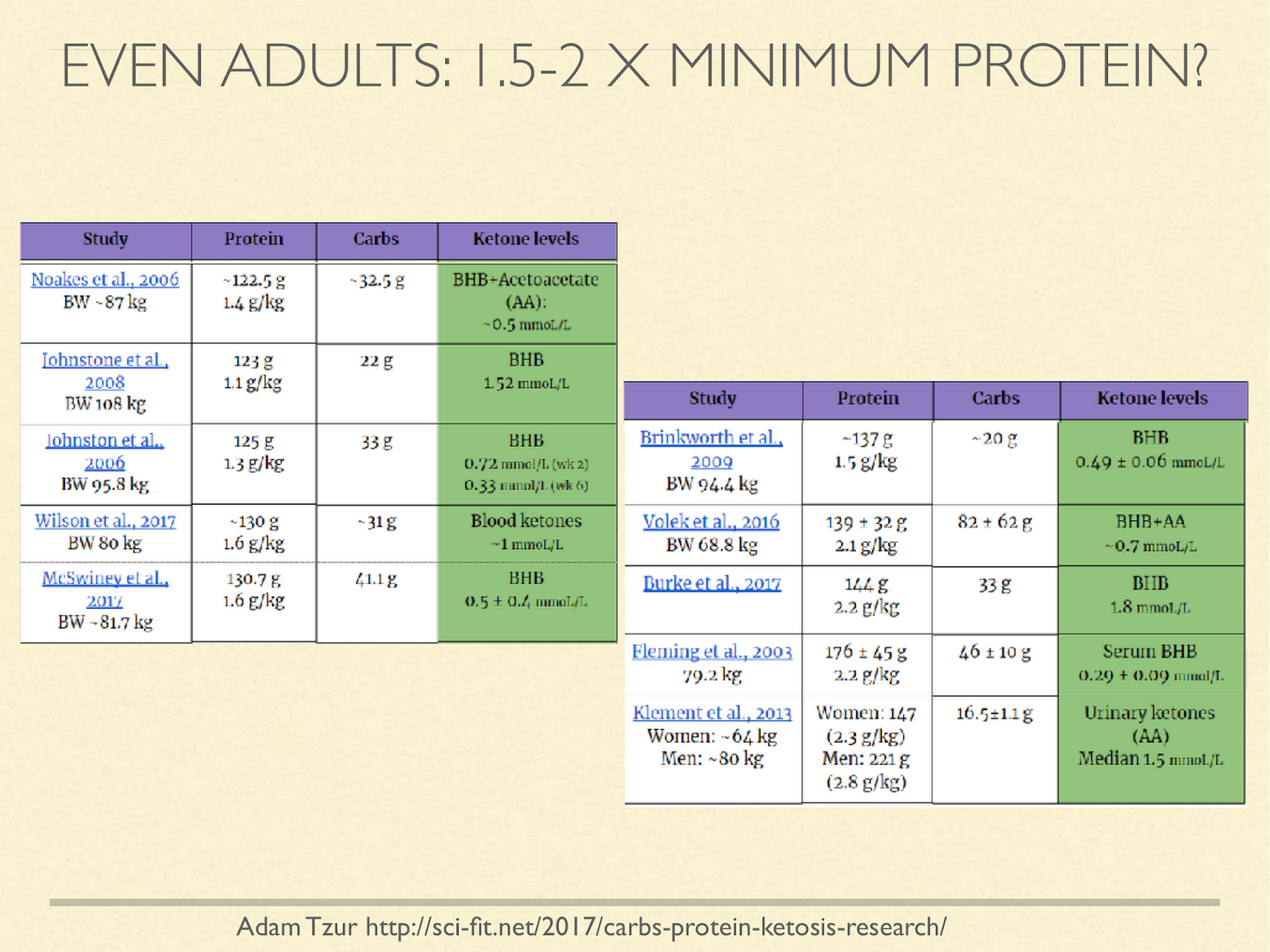
I don’t know of a study with the express purpose to find the upper bound of protein for ketosis, but we can look at studies that recorded it. Notice that the figures in this chart are using current weight, not ideal weight, and many of them are studies in overweight people, so the g/kg estimates look lower than they would be if using ideal weight. I’d love to see this question approached systematically, but the survey does at least suggest that protein levels above our minimum needs based on positive nitrogen balance still support ketosis.
[Graphic from: http://sci-fit.net/2017/carbs-protein-ketosis-research/ ]

When you compare adult humans with other species instead of with children, it’s even more impressive. Dogs are in many ways similar to humans. Our digestive anatomy and physiology is very similar. Dogs can reach ketosis from fasting, but it takes longer, and never attains the same level [Cra1941].
With adequate protein in the diet, it doesn’t happen to any significant degree at all [Rom1981], [Kro1973], [San2015]. I have spoken with staff at KetoPet Sanctuary, who treat cancerous dogs with ketosis.
They tell me that it is challenging to keep dogs in ketosis. They have to use a combination of protein restriction, calorie restriction, and MCT oils. It takes constant monitoring and adjustment.

Rodents are often used in experimental conditions, and I do think they are very useful models, but it takes more protein or calorie restriction to achieve an appropriate degree of ketosis than it would with humans. The line between adequate protein and too much for ketosis is almost vanishingly small [Stephen Phinney Q&A Low Carb Cruise 2017], and the levels they achieve are again much less spectacular [Benjamin Bikman, personal communication]. Similarly, almost any level of dietary carbohydrates is enough to shut down ketosis [Richard David Feinman, personal communication]. Some researchers believe this has to do with their relative lack of brains, since ketosis has been thought of as a way to spare glucose for the brain. But ketosis isn’t the only solution for that.

Obligate carnivores are always on very low carb diets, so you might think they are always in ketosis, but that’s not at all the case. In fact they are specialised at gluconeogenesis, that is, getting all their energy needs met by converting protein into glucose. Protein needs tend to be high. Cats have much higher protein needs than omnivores and surprisingly, they don’t adapt well to reduced protein or fasting [Cen2002]. They don’t seem to have good mechanisms to compensate for the various amino acid and vitamin deficiencies that develop, so they suffer from ammonia toxicity, methylation problems, and oxidative stress. They do produce ketones fasted, but they don’t seem to use them in a productive way and they actually accumulate fatty acids in the liver when fasted; the opposite of what humans do, because they are still producing glucose, they become like human type two diabetics.
Dolphins are particularly interesting because they have really large brains, and they eat a diet that would be expected to be ketogenic if fed to humans. However, they don’t seem to even generate ketone at all, not even when fasting. Instead, they ramp up gluconeogenesis [Rid2013]. They keep their bodies and their brains going by increased glucose.
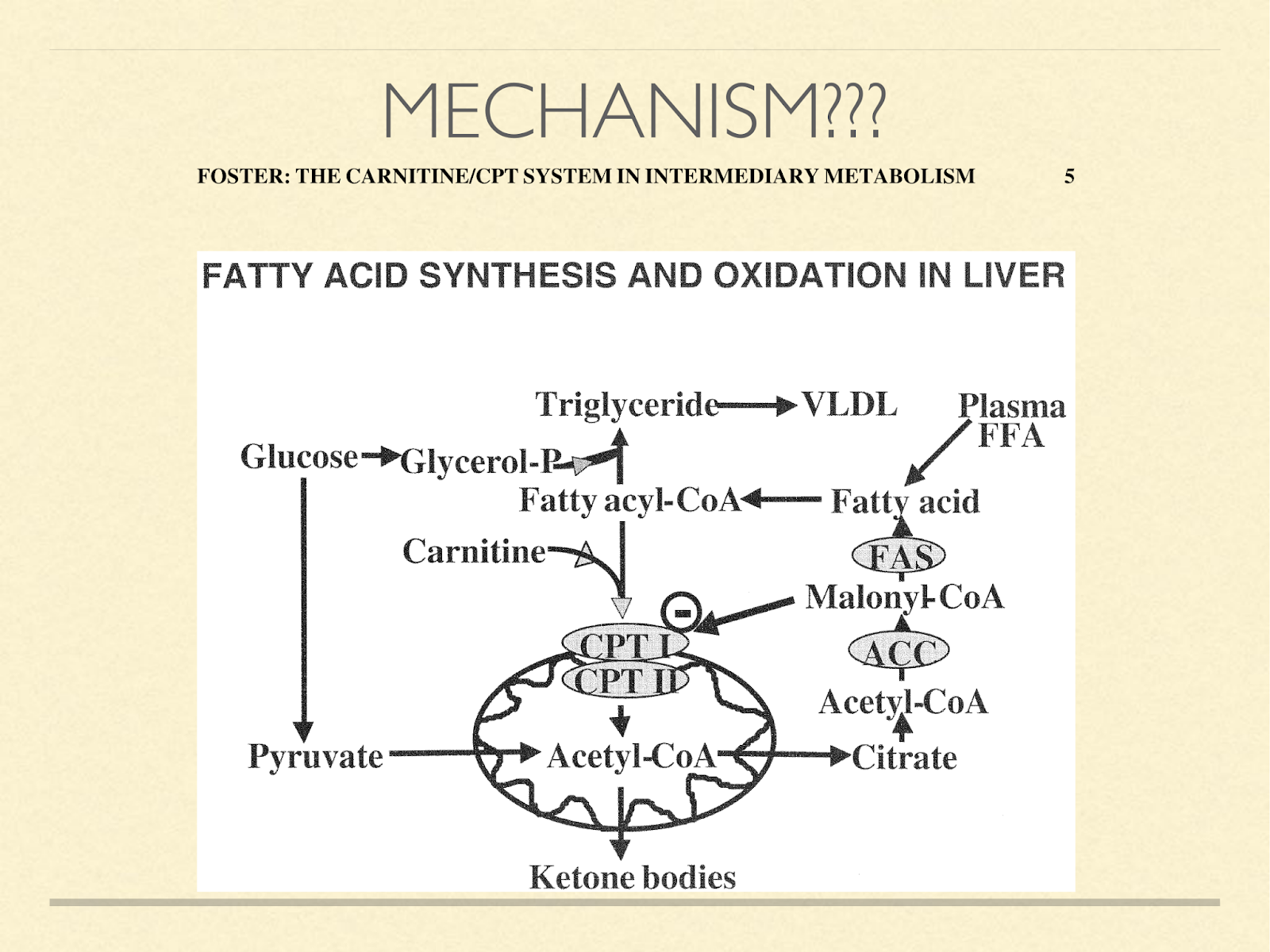
When faced with this observation that humans use ketosis even when they don’t have to for glucose production, one obviously wonders how this happens from a mechanistic standpoint. I have never seen the question raised in the literature, let alone answered. If I were to take a guess, I’d say it probably happens somewhere in this process.
CPT1A is a kind of gatekeeper, transporting fatty acids into the mitochondria for oxidation. This is normally a necessary step in the creation of ketone bodies. The coenzyme malonyl-CoA inhibits CPT1A [Fos2004]. The functional reason it does that is because malonyl-CoA is a direct result of glucose oxidation and is on the path to de novo lipogenesis. It could be inefficient to be both generating fat and oxidizing it. So this is a convenient signal to slow entry of fat into the mitochondria. However, its action is not stictly linear.
It uses hysteresis.
Hysteresis is a way of preventing thrashing back and forth between two states at the threshold of their switch. For example, if you set your thermostat to 20°C, you would not want the heater to be turned on when the temperature drops to 19.999 and turned off again at 20. This would result in constant switching.
Instead, a thermostat waits until the temperature drops a little lower before activating the heater, and heats it a little more than required before deactivating it. Hysteresis is implemented in CPT1A by its becoming insensitive to malonyl-CoA when levels of it are low [Ont1980], [Bre1981], [Gra1988], [Gre2009], [Akk2009].
That means that once CPT1A becomes very active in transporting fatty acids, it takes time before the presence of malonyl-CoA will inhibit CPT1A at full strength again. That means that fluxuations in glucose oxidation, or small, transient increases in glucose oxidation don’t disturb the burning of fatty acids or the production of ketones.
It could be the case that humans develop more insensitivity to malonyl-CoA under ketosis than other species do, allowing them to metabolise more protein without disturbing ketosis. Among humans, this is case in populations such as some Inuit with the Artic variant of CPT1A. That mutation slows down CPT1A activity immensely. This was permitted by their diet which was very high in polyunsaturated fats from sea mammals.
Polyunsaturated fats upregulate fatty acid oxidation by a large proportion compared to saturated fats [Cun2002], [Fra2003], [Fue2004], so this mutation would not necessarily have been disruptive of ketosis in that population when eating their natural diet [Lem2012].
But a second effect of the same gene further decreases the sensitivity of CPT1A to inhibition by malonyl-CoA. That means they are less likely to be knocked out of ketosis by high protein intake. I will go into this in much greater detail in my upcoming talk at AHS18.

The second question that comes to mind is what does this difference imply about our evolutionary environment?
I would suggest that for humans to have developed the ability to stay in ketosis even with more than sufficient protein intake, we must have at least have spent frequent long periods in a condition of very low carbohydrate, high fat access, either exogenously or endogenously, and more than adequate protein as a dietary norm.
Finally why?
Why do we stay in ketosis even when we have enough protein to feed the brain glucose without compromising lean mass. Or to put it another way:
Other animals continue to burn through lean mass with or without ketosis until they have enough protein to fuel everything with glucose. I suspect it has something to do with our brains. I’ll suggest a few hypotheses along these lines.
The next few slides summarise topics I’ve spoken and written about before. Please see Optimal Weaning from an Evolutionary Perspective for more details and links about brain growth and our acquired reliance on meat during evolution.
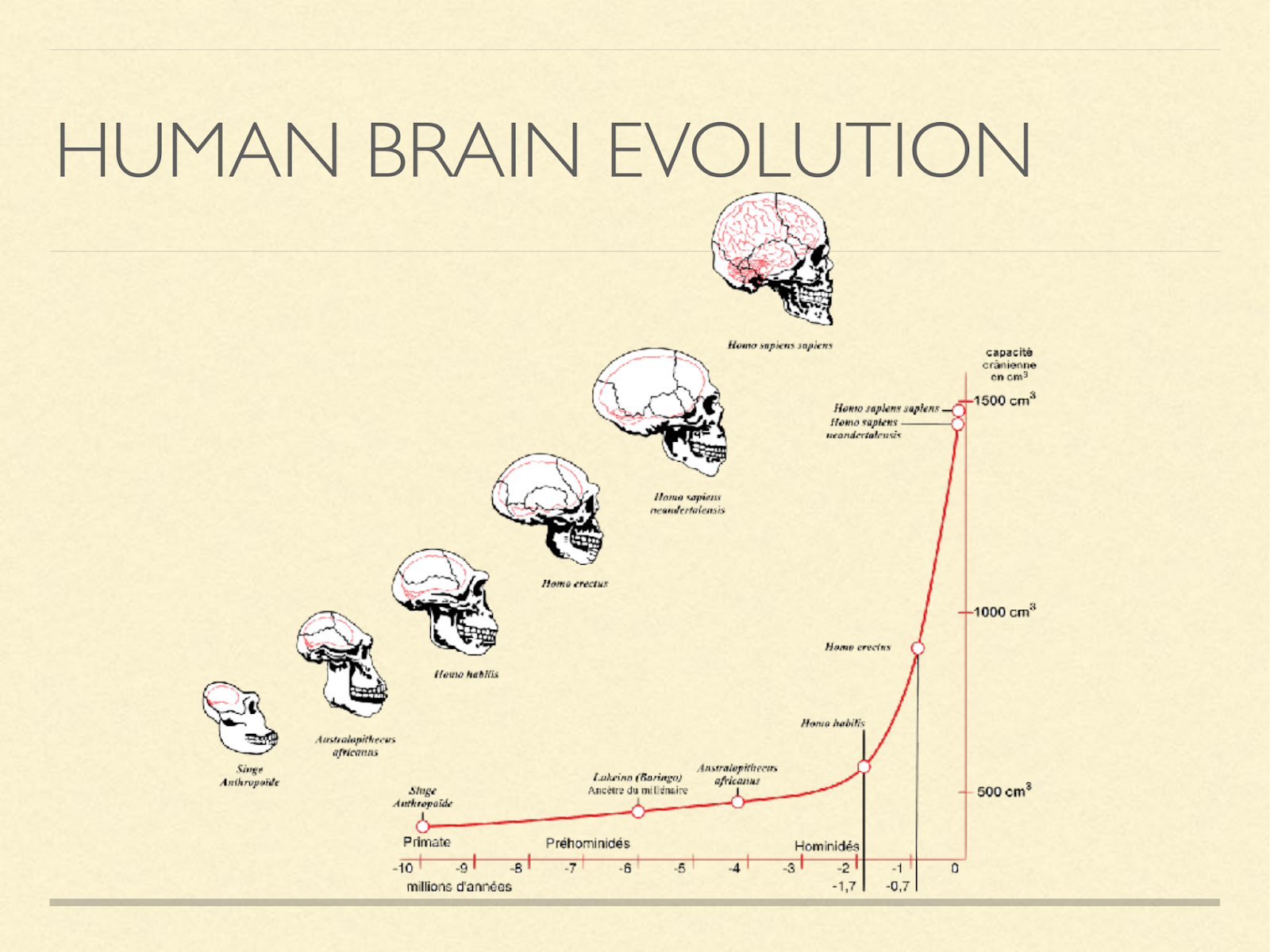
Our brains are big. Primates are already big brained for mammals, and from that starting point our brains tripled in size over the course of a couple million years.
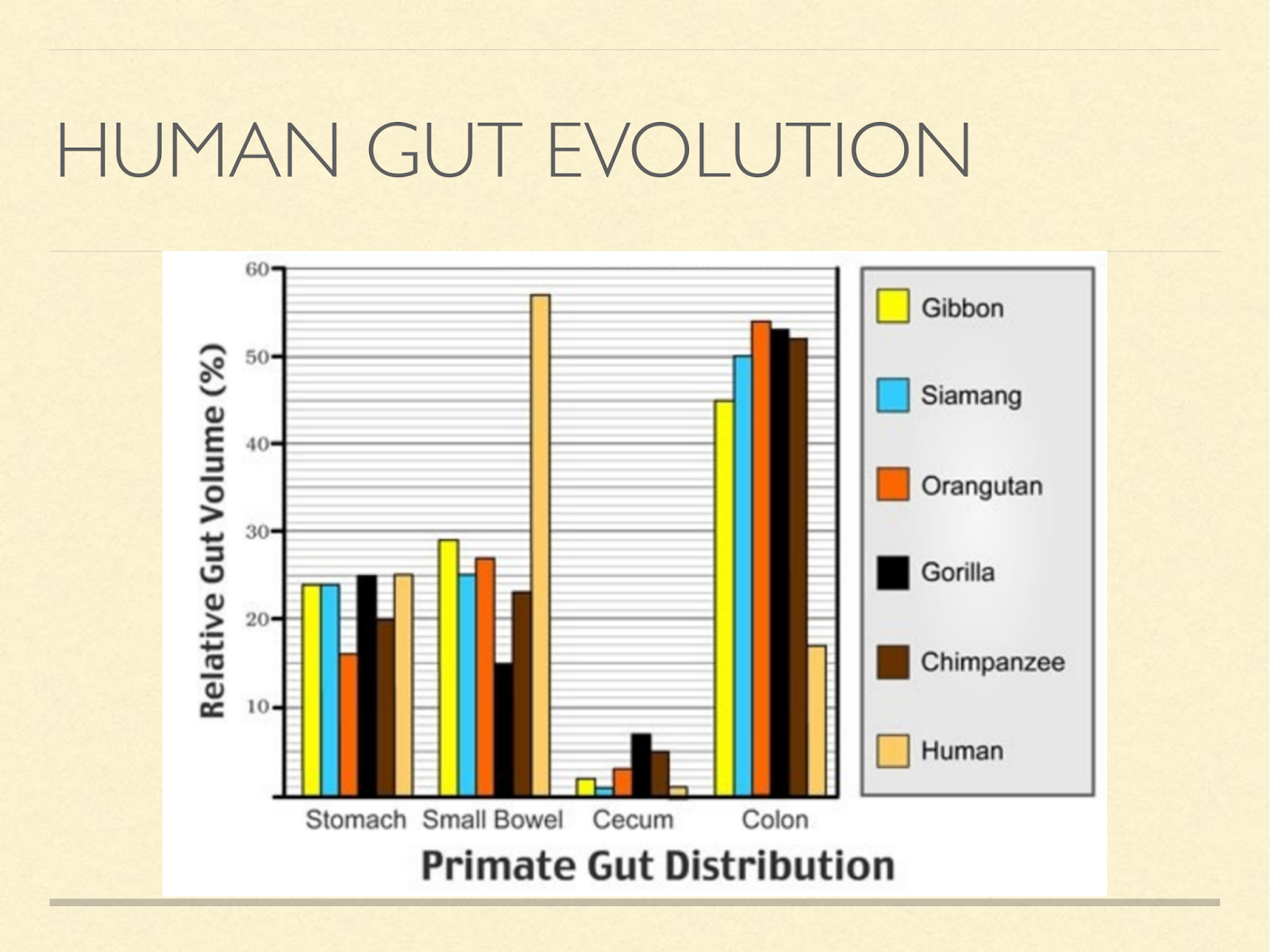
Brains take a lot of energy to run, to accommodate that we made a trade. Herbivores get most of their energy from fibre by fermenting it in the gut. But this isn’t very efficient, because intestines also take a lot of energy.
So we transferred to a strategy of eating fat directly giving up colon size for brain size.
To get enough fat directly, we had to eat meat. [Graphic from: Milton, Katharine. “Nutritional Characteristics of Wild Primate Foods: Do the Diets of Our Closest Living Relatives Have Lessons for Us?” Nutrition 15, no. 6 (June 1999): 488–98. https://doi.org/10.1016/S0899-9007(99)00078-7. version enhanced with colour by http://roarofwolverine.com/archives/219 ]

Energy is one reason we might want to stay in ketosis. Human brains use an extraordinary amount of energy, at least 20% in adults some 40g/day of that has to come from glucose, because it houses some of the few types of cells that are glucose bound.
But the rest can be met by ketones.
Our brains use ketones preferentially when they are available. Though in the modern context, that’s not very often.

If adult brains weren’t large and expensive to run enough, consider how much bigger the brain of a child is relative to the body. [Graphic from: http://vertpaleo.org/Society-News/Blog/Old-Bones-SVP-s-Blog/December-2013/Growing-up-(and-out,-and-sidways,-and-around).aspx ]
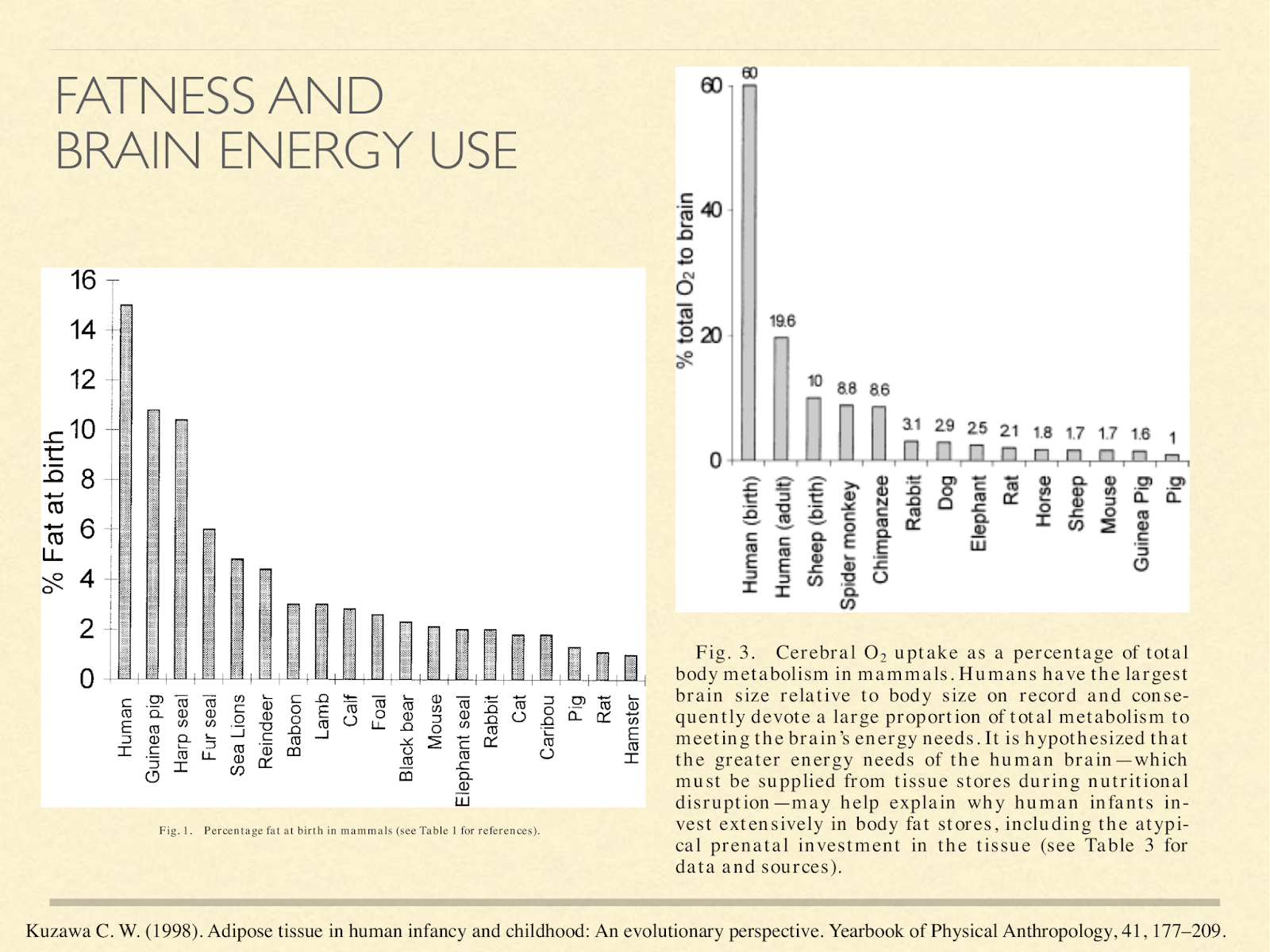
This may explain why human babies are so fat. These graphs are from a paper exploring different hypotheses about baby fat [Kuz1998], one of them being to supply the brain energy in the form of ketones. The one on the left shows % body fat at birth in different species.
Newborn humans come in at 15% fat. That actually gets higher in the first several months of life, peaking at about 25%. The only other primate in that graph is the baboon infant at 4%. The one on the right is what percent of oxygen metabolised by the whole body is going to the brain:
Humans at birth 60%, human adults 20%,… the adult chimpanzee comes in at about 9%.
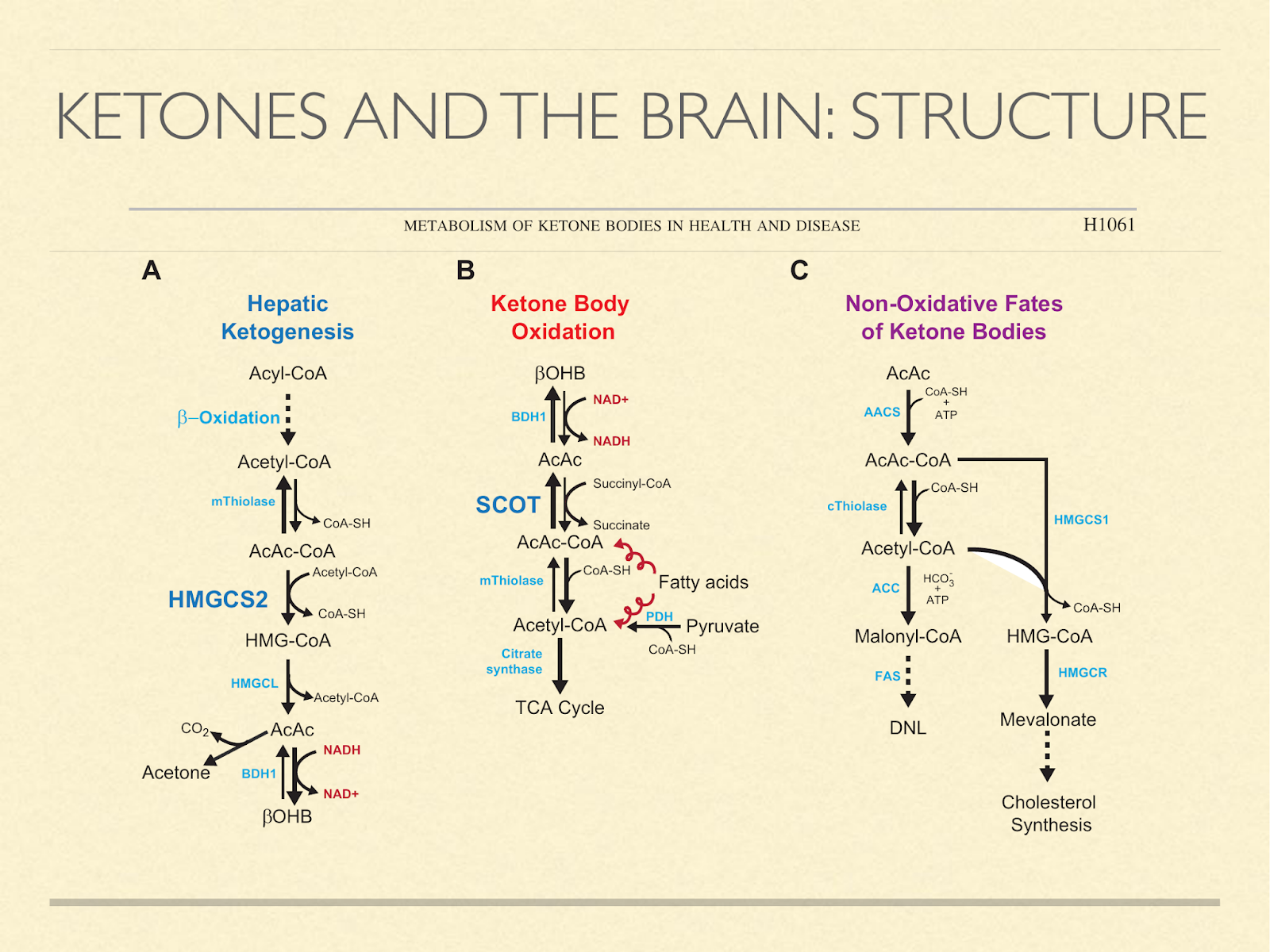
Another consideration is building materials, since our brains are made mostly of fat and cholesterol and we know that ketones are used to synthesize those in situ. The diagram here [Cot2013] shows pathways of how ketones can be generated, oxidized, or used to make fat and cholesterol.
Fetuses and newborns use ketone bodies extensively, as I mentioned previously. But the point here is that it’s not just because they’re using it for fuel. It’s also a source of structural components.
In light of that, it seems like a reasonable hypothesis that ketogenic capacity in humans is so pronounced in childhood because the brain is developing, and ketones are for some reason the preferred material.
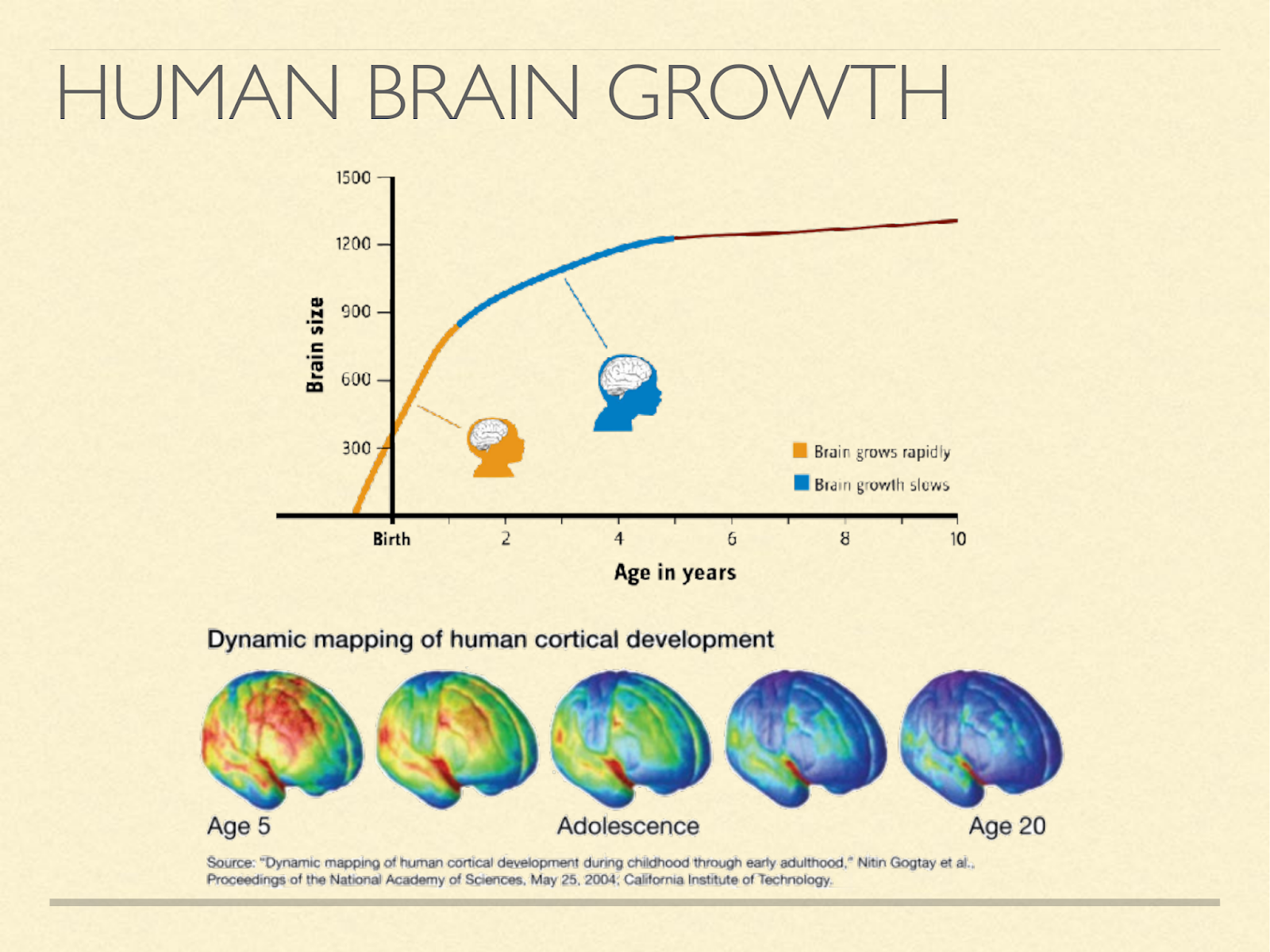
Other species tend to wean at the time when brain growth stops.
That means that for them ketogenesis stops at the same time brain growth stops. In humans brain growth doesn’t stop at weaning [Ken2005], [Mar1982], [Dob1973], [Dek1978]. Even after it reaches about full size in adolescence, it continues to change structurally well into adulthood. However, quantitatively, this structural cost is very small compared to energy considerations [Kuz1998], and so that hypothesis seems relatively weak on its own.

Another set of ideas comes from the metabolic effects we see in the lab and clinic. Some of the strongest, most consistent effects we’ve seen therapeutically from ketogenic diets take place in the brain, These are just a few metabolic changes relative to a high carb diet. Each can have profound effects on the workings of the brain.
I do want to draw attention to the last one about availability of arachadonic acid and DHA. These are important for the brain as they make up the phospholipids, and they are subject to a lot of turnover. Each of these effects has been proposed as a solution to the mystery of why a ketogenic diet treats epilepsy so effectively [Bou2007], [Nyl2009], [Mas2012], [deL2014].

But it’s not just epilepsy that ketosis is good for. Epilepsy is just the condition with the most research, and the widest acknowledgment. Other conditions for which at least some evidence supports improvement via a ketogenic diet
include neurological disabilities in cognition and motor control [Sta2012]; the benefit here may have to do with the proper maintenance of brain structures such as myelination (Recall phases: tear down damage, rebuild).
Survival after brain damage, the hypoxia of stroke or blows to the head is improved in animal models [Sta2012].
There is even animal evidence that brain damage due to nerve gas is largely mitigated by being in a state of ketosis during the insult [Lan2011]. Again, this suggests a structural support and resilience provided by a ketogenic metabolism. Resilience comes in part from not being as susceptible to damage in the first place, and that could be from reduced oxidative stress when using ketones for fuel. Ketogenic diets as a treatment for cancer are controversial, but some of the best evidence in support of it comes from glioblastomas. See e.g. [Zuc2010], [Sch2012].
This could be due mostly to the hypoglycemia stalling the rate of tumour development. And to venture into an area less well studied, but of critical importance given the epidemic that would be more apparent were it less taboo, there is preliminary evidence in the form of case studies that ketogenic diets may be promising treatments for many psychiatric illnesses too, for example, [Kra2009], [Phe2012].
Given that anticonvulsants are also used to treat bipolar, and the solid results of ketogenic diets on epilepsy, this may not be surprising. Additionally, the enhanced availability of AA and DHA may play a crucial role because these fatty acids are critical for the brain, and dysregulation in their flux has been associated with bipolar disorder and schizophrenia.
See e.g. [McN2008] and [Pee1996].

I would almost like to call a ketogenic diet a brain-growth mimicking diet.
The question of how and why humans are so ketosis prone may lead to interesting new insights about us as a species.
We seem to avoid giving up ketosis as long as possible.
only halting it when we take in so much glucose exogenously that we have to store it.
It seems likely that it facilitated the evolution of our brains,
that organ that makes us so different from other animals that we sometimes forget we are animals.
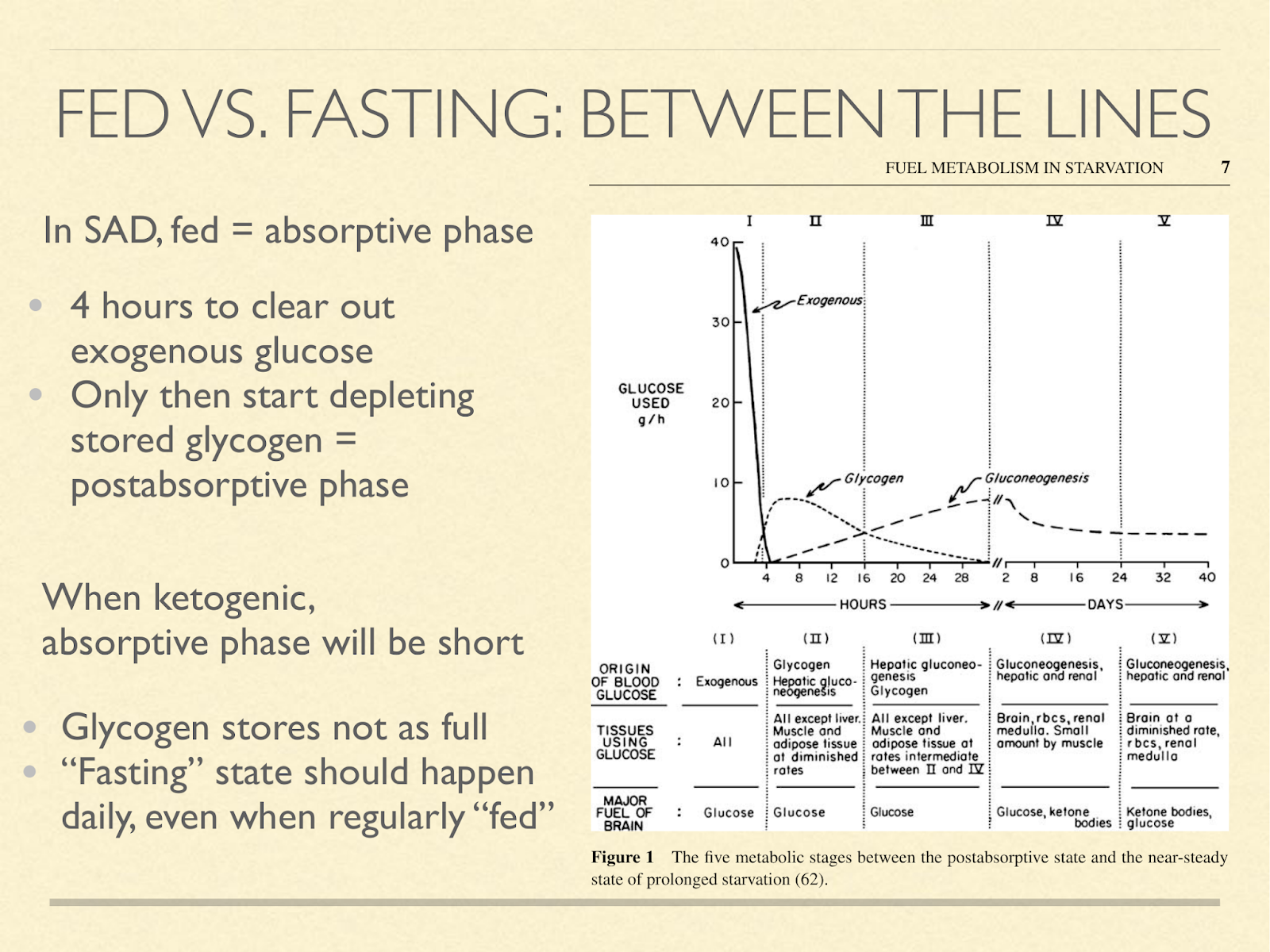
Returning to the importance of metabolic switching between glucose and ketone mode, there seems to be a false dichotomy. There is a stage that doesn’t usually come up in discussions of fed and fasted, and that’s the “postabsorptive” phase. The absorptive phase on a high carb diet lasts about 4 hours. That’s how long it takes to clear away the exogenous glucose. Only after that can you start the postabsorptive phase, marked by using glycogen as your source of blood sugar. Other than overnight, SAD dieters typically don’t go more than 4 hours without eating, and so we don’t get very far. But if you are on a protein and calorie sufficient very low carb diet, then even after eating, your glycogen stores don’t get that full in the first place. I don’t know how long it takes to get from the meal to maximum glycogen storage, but essentially, we should expect to get to a SAD dieter’s postabsorptive almost immediately after a meal, and easily into the ketogenic zone every day.
You can accentuate this by demanding more energy between meals (exercise) or eating less frequently, for example only once or twice a day. Interestingly, this often naturally happens to ketogenic dieters. ( Graphic from [Cah2006] )
<

On a high carb diet, you might need to fast to attain an enlightened brain state. On a ketogenic diet, as a human, that doesn’t appear to be necessary.
References
In the interest of time, I did not do my usual practice of end-to-end citations. I will probably return to fix that later!
[Ada1975] Adam, P. A., N. Räihä, E. L. Rahiala, and M. Kekomäki. “Oxidation of Glucose and D-B-OH-Butyrate by the Early Human Fetal Brain.” Acta Paediatrica Scandinavica 64, no. 1 (January 1975): 17–24.
[Akk2009] Akkaoui, Marie, Isabelle Cohen, Catherine Esnous, Véronique Lenoir, Martin Sournac, Jean Girard, and Carina Prip-Buus. “Modulation of the Hepatic Malonyl-CoA–carnitine Palmitoyltransferase 1A Partnership Creates a Metabolic Switch Allowing Oxidation of de Novo Fatty Acids1.” Biochemical Journal 420, no. 3 (June 15, 2009): 429–38. https://doi.org/10.1042/BJ20081932.
[Bou2007] Bough Kristopher J., and Rho Jong M. “Anticonvulsant Mechanisms of the Ketogenic Diet.” Epilepsia 48, no. 1 (January 4, 2007): 43–58. https://doi.org/10.1111/j.1528-1167.2007.00915.x.
[Bou1986] Bougneres PF, C Lemmel, P Ferré, and D M Bier. Ketone body transport in the human neonate and infant. J Clin Invest. 1986 Jan; 77(1): 42–48.
[Bre1981] Bremer, J. “The Effect of Fasting on the Activity of Liver Carnitine Palmitoyltransferase and Its Inhibition by Malonyl-CoA.” Biochimica Et Biophysica Acta 665, no. 3 (September 24, 1981): 628–31.
[Cah2006] Cahill, George F. “Fuel Metabolism in Starvation.” Annual Review of Nutrition 26, no. 1 (August 2006): 1–22. https://doi.org/10.1146/annurev.nutr.26.061505.111258.
[Cen2002] Center, S. Feline Hepatic Lipidosis. June 2002. Australian Veterinary Practitioner 32(2)
[Cot2013] Cotter, David G., Rebecca C. Schugar, and Peter A. Crawford. “Ketone Body Metabolism and Cardiovascular Disease.” American Journal of Physiology-Heart and Circulatory Physiology 304, no. 8 (April 15, 2013): H1060–76. https://doi.org/10.1152/ajpheart.00646.2012.
[Cra1941] Crandall, Lathan. A comparison of ketosis in man and dog. March 1, 1941 The Journal of Biological Chemistry. 138, 123-128.
[Cun2002] Cunnane, S. C., K. Musa, M. A. Ryan, S. Whiting, and D. D. Fraser. “Potential Role of Polyunsaturates in Seizure Protection Achieved with the Ketogenic Diet.” Prostaglandins, Leukotrienes, and Essential Fatty Acids 67, no. 2–3 (September 2002): 131–35.
[Cun2016] Cunnane SC, Courchesne-Loyer A, Vandenberghe C, et al. Can Ketones Help Rescue Brain Fuel Supply in Later Life? Implications for Cognitive Health during Aging and the Treatment of Alzheimer’s Disease. Frontiers in Molecular Neuroscience. 2016;9:53. doi:10.3389/fnmol.2016.00053.
[Dek1978] Dekaban AS. Changes in brain weights during the span of human life: relation of brain weights to body heights and body weights. Ann Neurol. 1978 Oct;4(4):345-56.
[deL2014] Lima, Patricia Azevedo de, Leticia Pereira de Brito Sampaio, and Nágila Raquel Teixeira Damasceno. “Neurobiochemical Mechanisms of a Ketogenic Diet in Refractory Epilepsy.” Clinics 69, no. 10 (October 2014): 699–705. https://doi.org/10.6061/clinics/2014(10)09.
[Dob1973] John Dobbing and Jean Sands. Quantitative growth and development of human brain. Arch Dis Child. 1973 Oct; 48(10): 757–767.
[Fos2004] Foster, Daniel W. “The Role of the Carnitine System in Human Metabolism.” Annals of the New York Academy of Sciences 1033, no. 1 (November 2004): 1–16. https://doi.org/10.1196/annals.1320.001.
[Fra2003] Fraser, D. D., S. Whiting, R. D. Andrew, E. A. Macdonald, K. Musa-Veloso, and S. C. Cunnane. “Elevated Polyunsaturated Fatty Acids in Blood Serum Obtained from Children on the Ketogenic Diet.” Neurology 60, no. 6 (March 25, 2003): 1026–29.
[Fue2004] Fuehrlein, Brian S., Michael S. Rutenberg, Jared N. Silver, Matthew W. Warren, Douglas W. Theriaque, Glen E. Duncan, Peter W. Stacpoole, and Mark L. Brantly. “Differential Metabolic Effects of Saturated versus Polyunsaturated Fats in Ketogenic Diets.” The Journal of Clinical Endocrinology and Metabolism 89, no. 4 (April 2004): 1641–45. https://doi.org/10.1210/jc.2003-031796.
[Gra1988] Grantham, B. D., and V. A. Zammit. “Role of Carnitine Palmitoyltransferase I in the Regulation of Hepatic Ketogenesis during the Onset and Reversal of Chronic Diabetes.” Biochemical Journal 249, no. 2 (January 15, 1988): 409–14. https://doi.org/10.1042/bj2490409.
[Gre2009] Greenberg, Cheryl R., Louise A. Dilling, G. Robert Thompson, Lorne E. Seargeant, James C. Haworth, Susan Phillips, Alicia Chan, et al. “The Paradox of the Carnitine Palmitoyltransferase Type Ia P479L Variant in Canadian Aboriginal Populations.” Molecular Genetics and Metabolism 96, no. 4 (April 2009): 201–7. https://doi.org/10.1016/j.ymgme.2008.12.018.
[Ken2005] Kennedy GE. From the ape’s dilemma to the weanling’s dilemma: early weaning and its evolutionary context. J Hum Evol. 2005 Feb;48(2):123-45. Epub 2005 Jan 18.
[Kos2013] Kossoff, Eric H., Mackenzie C. Cervenka, Bobbie J. Henry, Courtney A. Haney, and Zahava Turner. “A Decade of the Modified Atkins Diet (2003–2013): Results, Insights, and Future Directions.” Epilepsy & Behavior 29, no. 3 (December 2013): 437–42. https://doi.org/10.1016/j.yebeh.2013.09.032.
[Kra2009] Kraft, Bryan D., and Eric C. Westman. “Schizophrenia, Gluten, and Low-Carbohydrate, Ketogenic Diets: A Case Report and Review of the Literature.” Nutrition & Metabolism 6 (February 26, 2009): 10. https://doi.org/10.1186/1743-7075-6-10.
[Kra1974] Kraus H, Schlenker S, Schwedesky D. Developmental changes of cerebral ketone body utilization in human infants. Hoppe Seylers Z Physiol Chem. 1974 Feb;355(2):164-70.
[Kro1973] Kronfeld DS. Diet and the performance of racing sled dogs. J Am Vet Med Assoc. 1973 Mar 15;162(6):470-3.
[Kuz1998] Kuzawa, Christopher W. “Adipose Tissue in Human Infancy and Childhood: An Evolutionary Perspective.” American Journal of Physical Anthropology 107, no. S27 (January 1, 1998): 177–209. https://doi.org/10.1002/(SICI)1096-8644(1998)107:27+<177::AID-AJPA7>3.0.CO;2-B.
[Lan2011] Jeffrey L. Langston, Todd M. Myers Diet composition modifies the toxicity of repeated soman exposure in rats. Neurotoxicology. 2011 Jun;32(3):342-9. doi: 10.1016/j.neuro.2011.03.001. Epub 2011 Mar 17.
[Lem2012] Lemas, Dominick J., Howard W. Wiener, Diane M. O’Brien, Scarlett Hopkins, Kimber L. Stanhope, Peter J. Havel, David B. Allison, Jose R. Fernandez, Hemant K. Tiwari, and Bert B. Boyer. “Genetic Polymorphisms in Carnitine Palmitoyltransferase 1A Gene Are Associated with Variation in Body Composition and Fasting Lipid Traits in Yup’ik Eskimos.” Journal of Lipid Research 53, no. 1 (January 1, 2012): 175–84. https://doi.org/10.1194/jlr.P018952.
[Mar1982] Martin, Robert D. Human brain evolution in an ecological context. Fifty-second James Arthur lecture on the evolution of the human brain 1982
[Mas2012] Masino, Susan A., and Jong M. Rho. “Mechanisms of Ketogenic Diet Action.” In Jasper’s Basic Mechanisms of the Epilepsies, edited by Jeffrey L. Noebels, Massimo Avoli, Michael A. Rogawski, Richard W. Olsen, and Antonio V. Delgado-Escueta, 4th ed. Bethesda (MD): National Center for Biotechnology Information (US), 2012. http://www.ncbi.nlm.nih.gov/books/NBK98219/.
[Mat2018] Mattson, Mark P., Keelin Moehl, Nathaniel Ghena, Maggie Schmaedick, and Aiwu Cheng. “Intermittent Metabolic Switching, Neuroplasticity and Brain Health.” Nature Reviews Neuroscience 19, no. 2 (January 11, 2018): 63–80. https://doi.org/10.1038/nrn.2017.156.
[McN2008] McNamara, Robert K., Ronald Jandacek, Therese Rider, Patrick Tso, Kevin E. Stanford, Chang-Gyu Hahn, and Neil M. Richtand. “Deficits in Docosahexaenoic Acid and Associated Elevations in the Metabolism of Arachidonic Acid and Saturated Fatty Acids in the Postmortem Orbitofrontal Cortex of Patients with Bipolar Disorder.” Psychiatry Research 160, no. 3 (September 30, 2008): 285–99. https://doi.org/10.1016/j.psychres.2007.08.021.
[Mun2016] Muneta, Tetsua, Eri Kawaguchi, Yasushi Nagai, Momoyo Matsumoto, Koji Ebe, Hiroko Watanabe, Hiroshi Bando. “Ketone Body Elevation in Placenta, Umbilical Cord, Newborn and Mother in Normal Delivery.” Glycative Stress Research 2016; 3 (3): 133-140
[Nat2014] Nation, Judy, Maureen Humphrey, Mark MacKay, and Avihu Boneh. “Linear Growth of Children on a Ketogenic Diet: Does the Protein-to-Energy Ratio Matter?” Journal of Child Neurology 29, no. 11 (November 2014): 1496–1501. https://doi.org/10.1177/0883073813508222.
[Nyl2009] Nylen, Kirk, Sergei Likhodii, and W. McIntyre Burnham. “The Ketogenic Diet: Proposed Mechanisms of Action.” Neurotherapeutics 6, no. 2 (April 2009): 402–5. https://doi.org/10.1016/j.nurt.2009.01.021.
[Ont1980] Ontko, J. A., and M. L. Johns. “Evaluation of Malonyl-CoA in the Regulation of Long-Chain Fatty Acid Oxidation in the Liver. Evidence for an Unidentified Regulatory Component of the System.” Biochemical Journal 192, no. 3 (December 15, 1980): 959–62. https://doi.org/10.1042/bj1920959.
[Pee1996] Peet, M., J. D. Laugharne, J. Mellor, and C. N. Ramchand. “Essential Fatty Acid Deficiency in Erythrocyte Membranes from Chronic Schizophrenic Patients, and the Clinical Effects of Dietary Supplementation.” Prostaglandins, Leukotrienes, and Essential Fatty Acids 55, no. 1–2 (August 1996): 71–75.
[Per1966] Persson B, Gentz J. The pattern of blood lipids, glycerol and ketone bodies during neonatal period, infancy and childhood. Acta Paediatr Scand 1966 Jul;55(4):353-62
[Phe2012] James R. Phelps, Susan V. Siemers & Rif S. El-Mallakh The ketogenic diet for type II bipolar disorder. Neurocase: The Neural Basis of Cognition DOI: 10.1080/13554794.2012.690421
[Rid2013] Ridgway, S. H. (2013). A Mini Review of Dolphin Carbohydrate Metabolism and Suggestions for Future Research Using Exhaled Air. Frontiers in Endocrinology, 4, 152.
[Rom1981] Romsos DR , Palmer HJ , Muiruri KL , Bennink MR Influence of a low carbohydrate diet on performance of pregnant and lactating dogs. The Journal of Nutrition [01 Apr 1981, 111(4):678-689]
[San2015] Seizures in Dogs and Cats. Sean Sanders. John Wiley & Sons, Feb 9, 2015
[Sch2012] Scheck, Adrienne C., Mohammed G. Abdelwahab, Kathryn E. Fenton, and Phillip Stafford. “The Ketogenic Diet for the Treatment of Glioma: Insights from Genetic Profiling.” Epilepsy Research 100, no. 3 (July 2012): 327–37. https://doi.org/10.1016/j.eplepsyres.2011.09.022.
[Sha1985] Shambaugh GE 3rd. Ketone body metabolism in the mother and fetus. Fed Proc. 1985 Apr;44(7):2347-51.
[Sta2012] (1, 2) Stafstrom, Carl E., and Jong M. Rho. “The Ketogenic Diet as a Treatment Paradigm for Diverse Neurological Disorders.” Frontiers in Pharmacology 3 (April 9, 2012). https://doi.org/10.3389/fphar.2012.00059.
[Zuc2010] Zuccoli, Giulio, Norina Marcello, Anna Pisanello, Franco Servadei, Salvatore Vaccaro, Purna Mukherjee, and Thomas Seyfried. “Metabolic Management of Glioblastoma Multiforme Using Standard Therapy Together with a Restricted Ketogenic Diet: Case Report.” Nutrition & Metabolism 7, no. 1 (2010): 33. https://doi.org/10.1186/1743-7075-7-33.