Keto-adapted but no (low) ketones? Part II

Beyond urinary ketones
Several years ago I wrote about a common complaint to do with measuring urine ketones: sometimes they stop registering even when someone is on a strict low carb diet. This typically seems to happen when ketosis is mild — the strips just aren't sensitive enough, although I've seen reports of negative urine strips even with very high serum β-hydroxybutyrate (BOHB) on some occasions.
Here's an example from Twitter.
Glucose: 67
— Rich Collins (@richcollins) February 11, 2020
Blood BHB: 4.3
Urinary Ketones: 0
Guess I’m keto adapted.
Makes you question those Inuit studies claiming they weren’t in ketosis based on urinary ketones.
(Also, about those Inuit and urinary ketones,

Rich is definitely on to something [OHe2018].)
Back when I wrote that blog post, very few keto dieters used blood test strips to measure ketosis, though. It was simply too expensive, with strips selling for $5 a pop! Now that you can get a blood ketone reading for a dollar or less, more people use this method, but the issue has reared itself again, albeit for different reasons. In this post, I'll explore some hypotheses to explain reduced blood ketones in certain people, despite obvious keto-adaptation.
Abbreviations:
- AcAc — acetoacetate
- BOHB — β-hydroxybutyrate
- BrAce — breath acetone
- ECT — electron transport chain
- FFAs — free fatty acids
- KB — ketone body
- LMHR — Lean mass hyper-responder
- MCR — metabolic clearance rate
- RQ — respiratory quotient
Contents
- Keto-adapted with low BOHB? Wut?
- Do lower BOHB levels reflect greater use?
- Acetoacetate is the proto-ketone-body
- β-hydroxybutyrate as an acetoacetate reserve
- What do we use acetoacetate for?
- AcAc:BOHB ratio as a marker of redox state
- Fat access at rest
- Footnotes
- End-to-end citations
Keto-adapted with low BOHB? Wut?
Saying you are keto-adapted when you aren't even showing high levels of BOHB in the blood almost seems like a contradiction, because ketosis is often defined as reaching a certain threshold of serum BOHB (typically 0.5 mmol) [1]. Yet this low BOHB (at rest, not during or post-exercise, which we'll consider separately) is not uncommonly reported with people who are very active (see below). An understandable assumption when your ketones are low is that you just aren't in ketosis, or at least not deep ketosis. However, this comes back to the "multi-variable" problem that we've talked about many times on this blog: When you have a system of production, level, and use, you can't determine all of them just by knowing one. Specifically, if what you want to know is how much of your energy you are getting from ketones, you can't tell that by the level in your blood, unless you also know how much is being produced (and excreted without being used).
So the question becomes, how can you tell whether the lower BOHB reflects lower use of ketones or not? We will consider some ways, but first, we need to have a clear understanding of the role of BOHB in the context of keto-adaptation.
Do lower BOHB levels reflect greater use?
One idea I've heard about why some athletic types are finding relatively lower levels of BOHB even though they consume what should be ketogenically low levels of carbohydrates and seem to be very well keto-adapted by other measures [2], is that because they exercise a lot, they take up ketones more readily for energy.
There are some problems with this theory. For example, this conjecture by itself is prone to the "knew it all along" fallacy [3]. Some of the same people who argue that BOHB is low in athletes because they use it efficiently also explain the finding that glucose levels are sometimes mildly elevated in the same population by saying that the function is to keep the fuel in ready. We see that what one would predict if one didn't know the answer might be the opposite of what we would say makes perfect sense after the fact. Ultimately we need a falsifiable hypothesis, not a "just so story".
Relatedly, production, levels, and use make up a three-part system that we would expect to fall into a steady state. If use is higher and levels are constant, then production must be higher, but the levels at this steady state don't have to be related to those at all, so long as they equilibrate. The only sensible relationship I can think of is that there needs to be enough to form a buffer given variable demand and lagging production rates. This is the essence of the glucose argument above, but it does not lend any insight to lower BOHB.
A more likely explanation to me would be that there is something about being either "metabolically healthy" or lean and aerobically fit (which typically coincide) that changes the steady state after adaptation. We'll get to that in a subsequent section.
First we'll dive into how ketones are used in order to try to eliminate the more-use hypothesis.
Acetoacetate is the proto-ketone-body
With all the research emphasis on BOHB, it is easy to forget that AcAc is actually the central ketone body [Whi2011]. It is the "parent", because both acetone and BOHB are derived from it. It is also the form that is used for energy and for synthesis of fat or cholesterol. BOHB has to be reversed back to AcAc before it can be used. You can see all of these pathways in figure 1, from Cotter et al. 2013 [Cot2013].
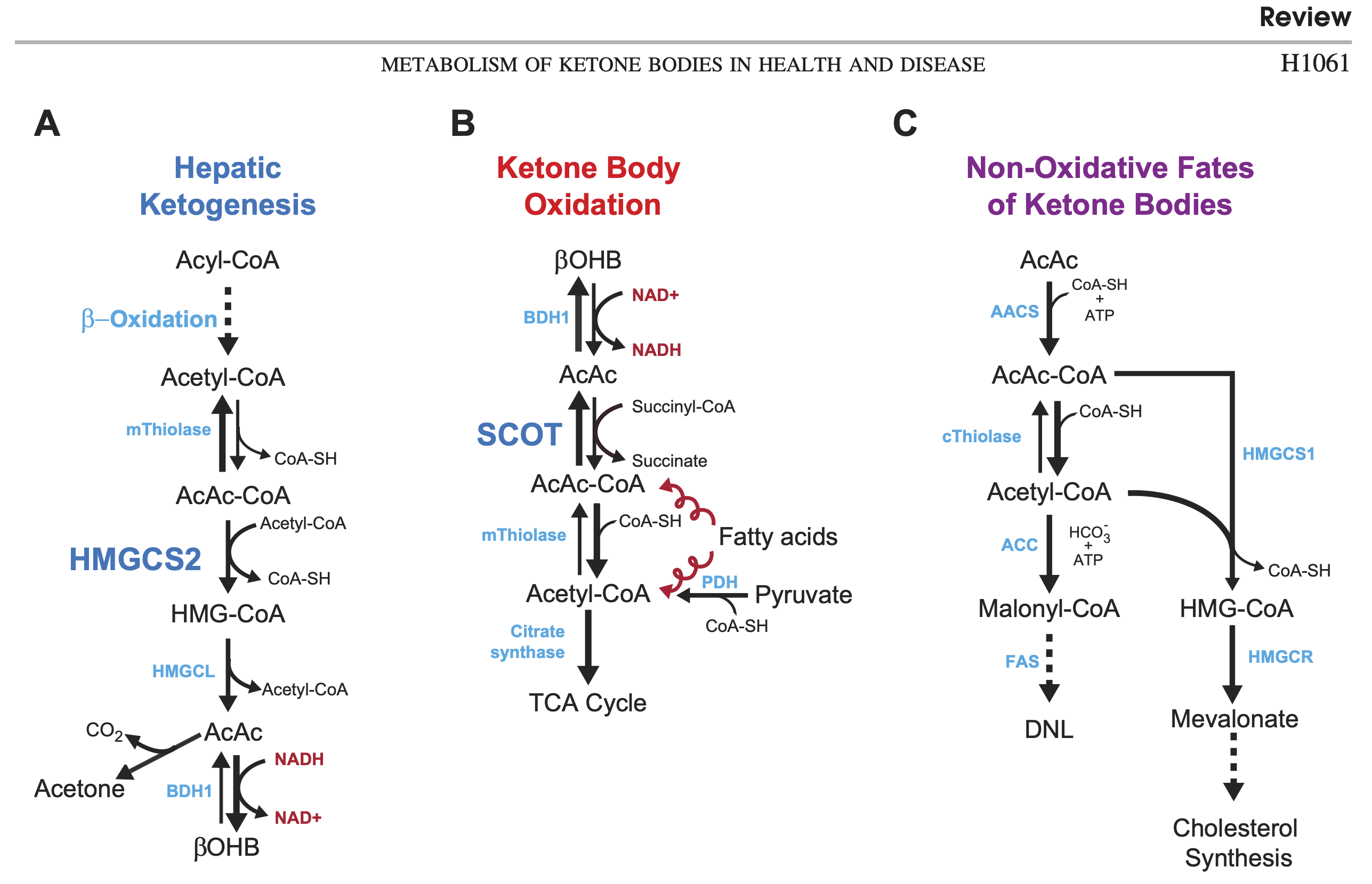
Acetoacetate is "unstable"
Notice that the production of acetone from AcAc just happens without requiring energy (spontaneous decarboxylation). This "instability" has been proposed as a functional reason AcAc largely gets converted to BOHB in the keto-adapted: to keep it from degrading. In other words, BOHB can be seen as a "storage" or "reserve" form of ketone body [Mus2002].
Is acetone a "waste product"?
Because this conversion is spontaneous, making AcAc unstable, this could be interpreted as a kind of unfortunate biochemical accident. I am reluctant to write it off this way, because many important metabolites have erroneously been assumed to be waste in the past, including AcAc and BOHB!
I don't have a proposal for a singular important function of acetone, but I think it is prudent to keep in mind that there might be one. Although it appears to be of minor quantitative relevance, acetone can be a substrate for gluconeogenesis in other animals. Reichard et al. surmised in 1979 that it may contribute perhaps as much as 11% of glucose synthesis in fasted humans, accounting for as much as 59% of the acetone produced [Rei1979]. That's significant.
Later, Kalapos wrote that because metabolic routes for acetone have been described, it "cannot further be regarded as a waste product of metabolism" [Kal1999]. He futher describes potential roles in peripheral fuel and pH regulation, which may be of critical importance.
β-hydroxybutyrate as an acetoacetate reserve
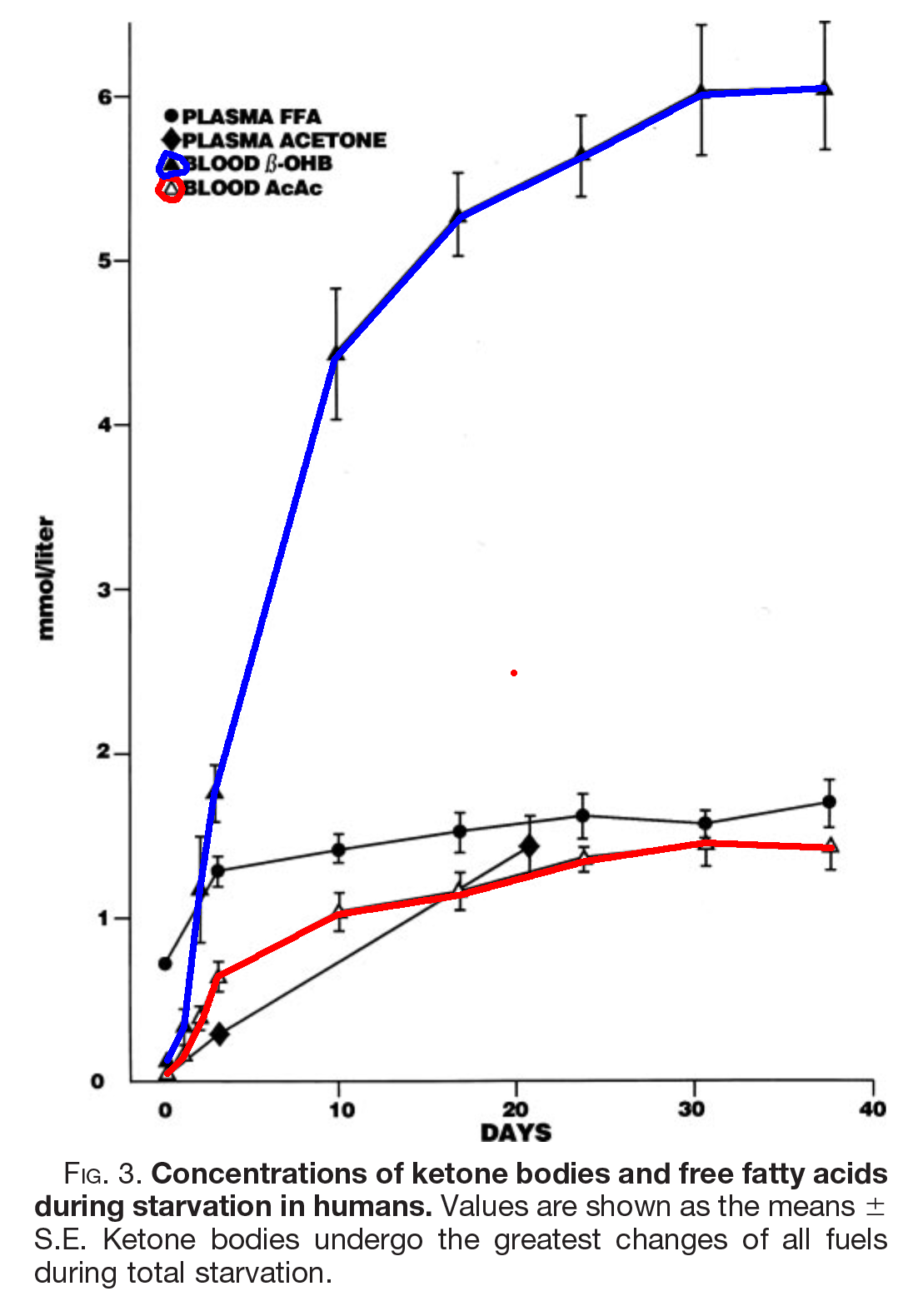
The above image, from [Owe2005], shows what happens to ketone bodies (KBs) in starvation, but the same happens on a ketogenic diet, as you can see here from [Nea2009]
In these studies of fasting and fed ketosis, AcAc tops out just over a mmol. BOHB continues to rise until it reaches just over 4 mmol in therapeutic ketogenesis, or 6 in fasting.
What appears to happen is that in the steady state, AcAc above and beyond 1.2 mmol or so is converted to BOHB.
So, one could certainly argue that what makes you keto-adapted is not the level of BOHB, but the level of AcAc, because BOHB only reflects overage. Insofar as this is true, acetone, as a proxy for AcAc, may be a better indicator of ketosis than BOHB. See footnotes for how to measure acetone [4].
What do we use acetoacetate for?
There are at least two important uses for AcAc that one might expect to lead to low BOHB even when you are generating a lot of AcAc: direct use for energy and for lipogenesis, especially cholesterol synthesis (cholesterol is a lipid).
Energy in the keto-adapted
If you are what Dave Feldman has dubbed a "lean mass hyper-responder" (LMHR), then when you are on a ketogenic diet, you often will have high levels of cholesterol carried in LDL and HDL, and low levels of triglycerides in those same particles. As the name suggests, these people tend to be lean and athletic. (Note that this high LDL effect may be reversed with high fat intake, possibly requiring high caloric intake. See Feldman's Cholesterol Drop protocol at the same site, but see also results from Paleomedicinawhich uses a very high fat but not hypercaloric protocol and also appears to "normalise" LDL. I expect, the low BOHB effect will be reversed as well, depending on protein intake.)
Feldman's theory about why this is, is that VLDL secretion is upregulated to deliver triglycerides for energy, but those triglycerides are then depleted quickly from VLDL particles, leaving LDL particles that still contain cholesterol. LDL is just VLDL after most of the triglycerides are taken up, and it has a relatively longer lifespan as a particle carrying the cholesterol remaining in it. In other words, according to the model, when we see high LDL in these cases, we're just seeing the empty traces of enhanced energy delivery. So this suggests that perhaps cholesterol synthesis goes up in order to make up VLDL particles sent out for energy needs.
Acetoacetate for cholesterol synthesis
As it turns out, AcAc appears to be the preferred material for cholesterol synthesis, not just in the brain as I've discussed previously (for example in this talk on ketosis and weaning), but also in the liver [Gee1983], [Ber1984].
Here is a diagram from [End1982] showing some different fates of AcAc in the liver. There is a similar one in [Gib1986].
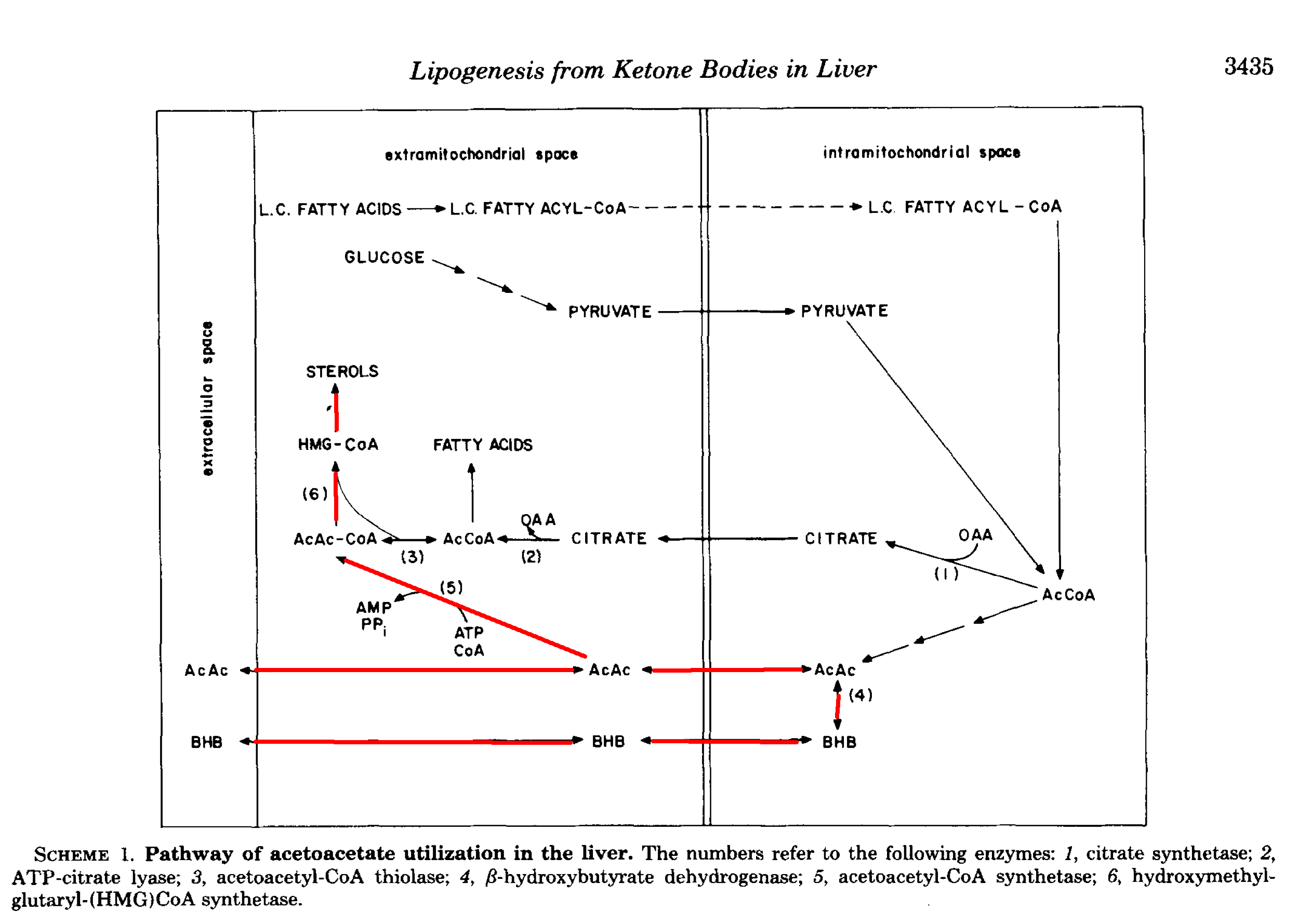
In a more recent review, Puchalska et al. report that up to 75% of cholesterol synthesis could come from AcAc, along with up to half of other newly synthesised lipids [Puc2017]. As they point out, it may seem odd—a kind of futile cycling—to take free fatty acids, oxidise them, and then turn them back into fat, but sometimes one form is better than another.
Specifically, the desired form may be the form of fat that can travel in lipoproteins along with cholesterol. As Salam et al. argue, the fact that free fatty acids specifically stimulate several enzymes in the the hepatic cholesterol synthesis pathway probably indicates a need to form the structural components of VLDL exactly in order to transport triglycerides out [Sal1989]. (They also note that the more saturated the fat, the more cholesterol per triglyceride was secreted.)
Why are babies only in mild ketosis despite having an immense amount of body fat? Perhaps because they are turning it into cholesterol not just in the brain, but everywhere they are making new cells. And since they are actually using that cholesterol that could explain why their LDL is low.
This might lead one to consider a revised explanation of Feldman's Energy Model, the Cholesterol Drop phenomenon, and its relationship to ketosis. Could the LMHR be making more cholesterol in order to transport energy, and this is using up AcAc resulting in lower ketones?
Appealing as the idea is it appears to be wrong. It turns out that fasting, and presumably also the fed ketogenic condition (as long as fat intake is not very high), while raising LDL, also reduces cholesterol synthesis [Bro2010], [Cre2018], [5]. At the same time, with the same underlying mechanism, it inhibits PCSK9, which regulates LDL receptors making them take up more. Both of these factors would be expected to lower, not raise cholesterol, so the higher LDL is more likely to be an effect of recycling, which high HDL would support.
It may be that in the context of high fat demand compared to intake cholesterol synthesis is downregulated to spare lipids for energy, while cholesterol recycling is increased, but the overall effect is still higher circulation. By the same reasoning, ketogenesis may likewise be deprioritised when fat is at a premium and energy for muscles is required. In this scenario, to preserve brain function, glucose may need to fill more of the gap.
Acetoacetate for energy
But triglycerides aren't the only way to get energy on a low carb diet. AcAc is another source of energy that can be used directly by muscles. It seems a reasonable hypothesis that those same LMHRs might use up AcAc directly more quickly, and thus have less leftover AcAc that needs to be converted into BOHB reserves. This might explain why BOHB levels tend to be relatively low in ketogenic athletes.
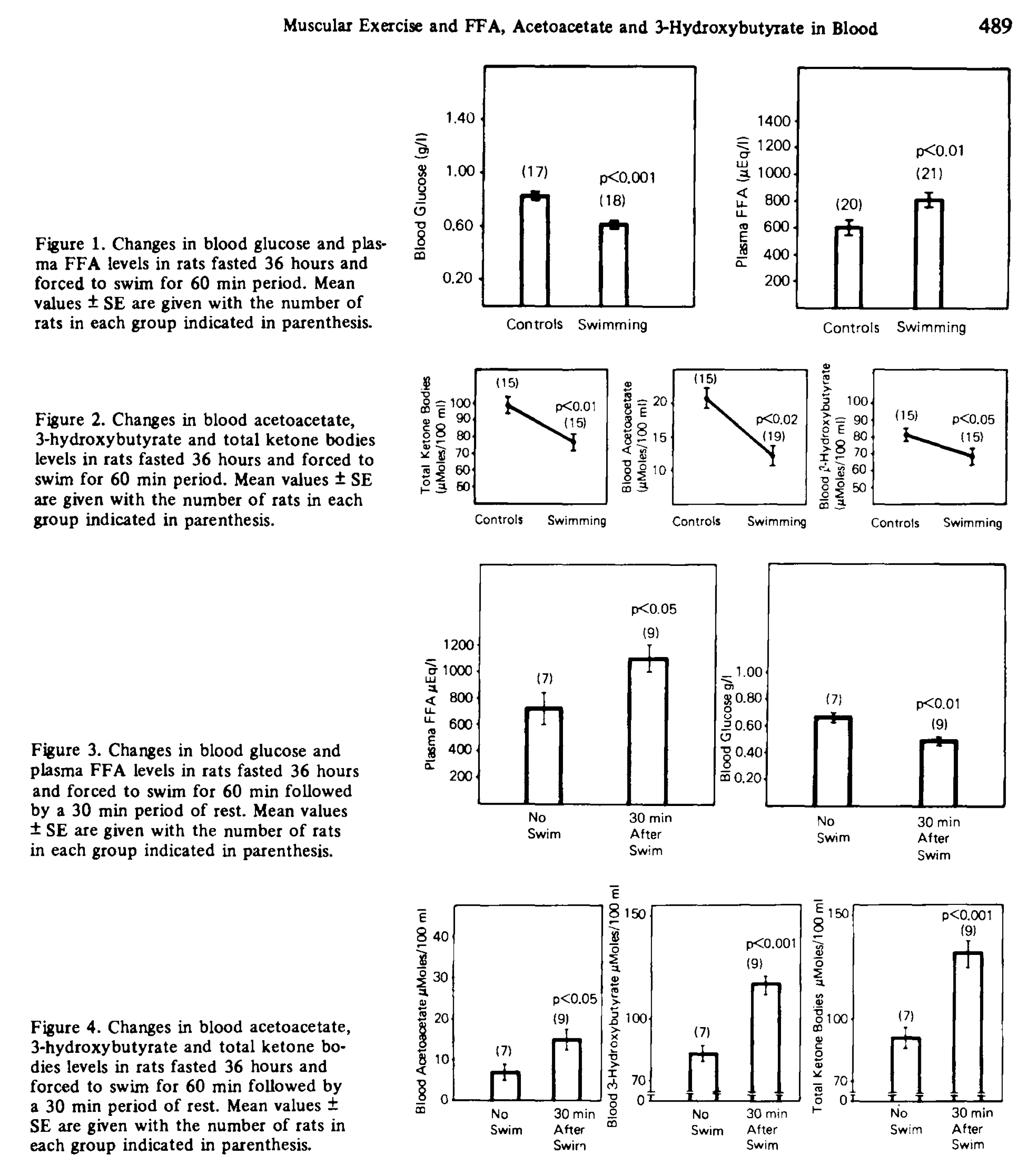
There is a rat study that seems to support this [Bon1974]. Rats were fasted for 36 hours, and then some of them were compelled to swim for 60 minutes. Swimmers had lower AcAc and BOHB than controls during the swim, and higher of both after, supporting peripheral uptake during exercise.
On the other hand, at least one study of keto-adapted humans paints a very different picture.
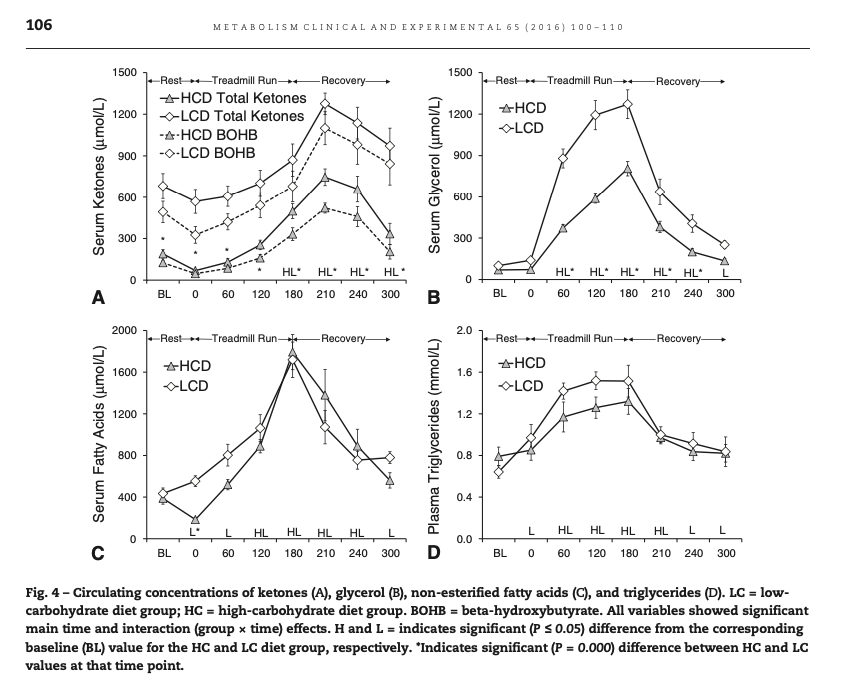
In this diagram from Volek at al. [Vol2016], we see that the low carb athletes had baseline levels of AcAc of about 0.2 mmol, and BOHB of about 0.5 mmol. During exercise the AcAc didn't change, but BOHB went up acutely. After exercise the elevated ketones fell off only slowly, unlike the elevated free fatty acids.
If AcAc were being used as energy, and BOHB were a reserve, we would not expect BOHB to increase during exercise as it did both absolutely and relative to AcAc.
Instead, this experiment supports the widely accepted idea that keto-adaptation entails a downregulation of ketone use in muscle tissue in order to spare it for the brain [Vol2011].
So which is it? The answer may depend on your ketosis level when you started.
Metabolic clearance rate (MCR) depends on baseline ketosis
As covered in the extensive review by Evans et al. [Eva2017], several factors affect the ability of skeletal muscle to oxidise KBs and MCR, but the degree of ketonemia is a crucial one. MCR drops off apparently exponentially as concentration rises, as does the effect of exercise of increasing MCR. This is shown in experiments from Hagenfeldt and Wahren [Hag1971]. As you can see, the higher the starting levels of ketosis, the less clearance there is.
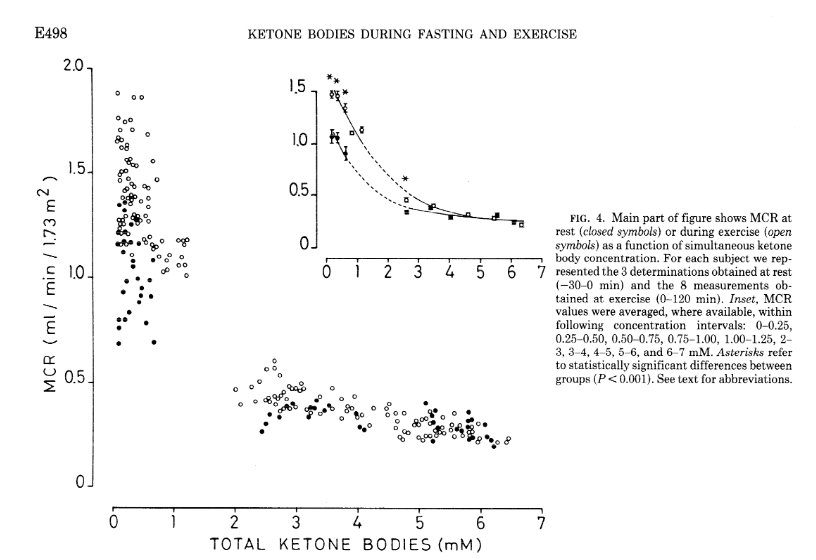
Notably, the concentration at the inflection point appears to fall in the 1-2 mmol range (2-3 for exercise effect), which may have implications for the definition of keto-adapted in the first place. The study is tantalisingly missing data within the 1-2 mmol range!
In general, we know that untrained animals tend to have higher post-exercise ketosis than trained ones (see e.g. [Bea1985]). So the fact that the athletes in Volek et al.'s study had rising KBs during exercise and falling after looks like it could simply be an effect of increased MCR from training. It's also of note that the total KBs they had at rest were only about 0.7.
This leaves us in a bit of a circle. Are the keto-adapted athletes showing lower KBs at rest because they have higher MCR from being trained athletes? If so, would that mean they require even higher levels of ketosis before the MCR effect would switch to ketone sparing mode? This would seem terribly maladaptive, since, as Evans et al. put it, "This [attenuation of exercise-stimulated MCR] is critical in the starvation response because the capacity of the liver to produce KBs closely matches the requirements of the brain to utilise KBs as an energy source."
Or is the fact that they start with lower KBs due to something else, in which case we're no closer to an answer. Maybe they both have a common cause.
AcAc:BOHB ratio as a marker of redox state
Recall that in Volek et al. as the KBs rise during exercise, all of the rise is in BOHB. Because AcAc is the parent KB, this means that as more KBs are produced, all of the new AcAc is either being used or converted to BOHB. Either way, the ratio of AcAc:BOHB is dropping from roughly 30% to roughly 20%. From a purely ratio perspective, this would look like the level increase from fasting going from maybe day 3 to day 7 in the Owens figure, even though the absolute levels are lower [6].
A primary determinant of AcAc:BOHB ratio is held to be the proportion of oxidised vs reduced forms of NAD, NAD+ and NADH respectively [Whi2011]. This is simply because the interconversion of KBs entails the conversion of NAD: AcAc + NADH ⟷ BOHB + NAD+.
The ratio of NAD+:NADH is often considered a marker of available energy. That's because electrons from NADH are used in the electron transport chain (ECT) in mitochondria to generate energy, leaving NAD+. However, there is another source of electrons that can be used as an alternative: FADH2. NADH goes through Complex I, and FADH2 goes through Complex II. From there the the electrons join the same pool.
The dominance of NADH/Complex I for energy is in part a carbohydrate metabolism phenomenon. When glucose is the primary source of energy, about five times as much NADH is provided to the ECT as FADH2, Whereas when fat is the primary source of energy, that ratio is only about twice as much. Please read Peter "Hyperlipid" Dobromylskyj for more.
So if you're thinking about carbohydrate metabolism by default, high NAD+:NADH can indeed indicate a shortage of energy. This is similar from a signalling point of view to the AMP:ATP ratio, which is reflected in AMPK activity. Like high AMPK, relatively high NAD+:NADH can also just indicate that fat metabolism is predominant. Conditions associated with high NAD+:NADH include fasting, caloric restriction, and exercise. These conditions also tend to result in high BOHB:AcAc, consistent with the equation.
What's actually happening with NAD ratios during exercise is a bit elusive. Ultimately, as described in an extensive review from White and Schenk [Whi2012], "[t]here are conflicting results in both animal and human studies as to whether or not exercise increases or decreases NAD+, NADH, and the NAD+/NADH ratio. There are many reasons that may underlie these differences, including training state, intensity of contraction, duration of exercise, time point of measurement during exercise, the analytical technique used to measure NAD+(H) and the NAD+/NADH ratio (e.g., fluorometric, biochemical, and MIM method), and the compartment that was measured (whole tissue, mitochondrial, or cytosolic)."
Oddly, this paper is frequently cited to support the claim that NAD+ is increased during exercise. That is not my reading of this. Instead those claims appear to be applicable to long-term adaptation. Interestingly, the long-term adaptation that may be most important is mitochondrial biogenesis.
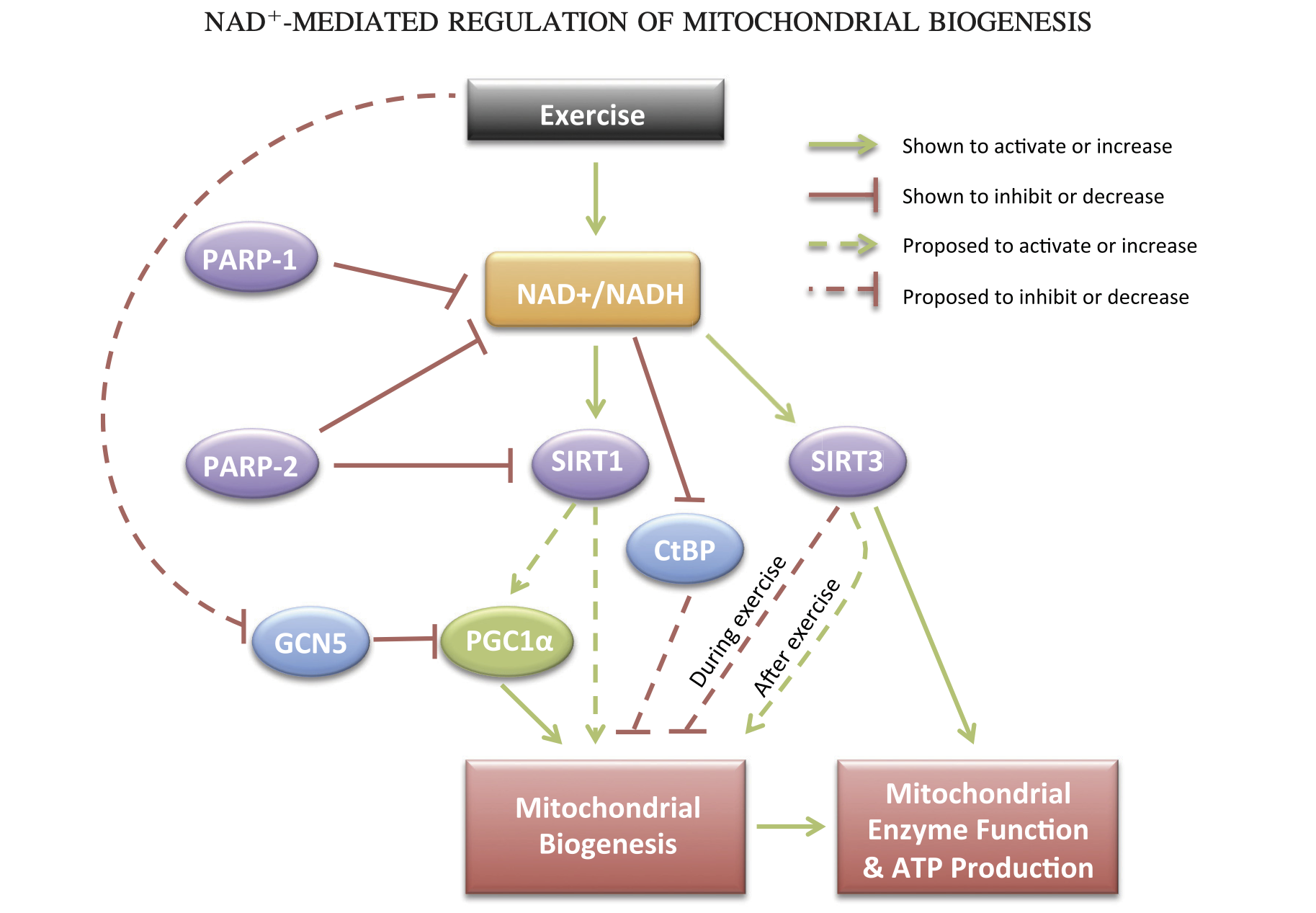
This adaptation makes perfect sense from an energy perspective. If an energy shortage is perceived — if we aren't getting enough ATP from the mitochondria we have, then we need to make more! But how would more mitochondria help if we have a global relative shortage of NADH? I think the answer might be due to compartmental differences. It may simply be part of the work of being a mitochondrion to keep more NADH.
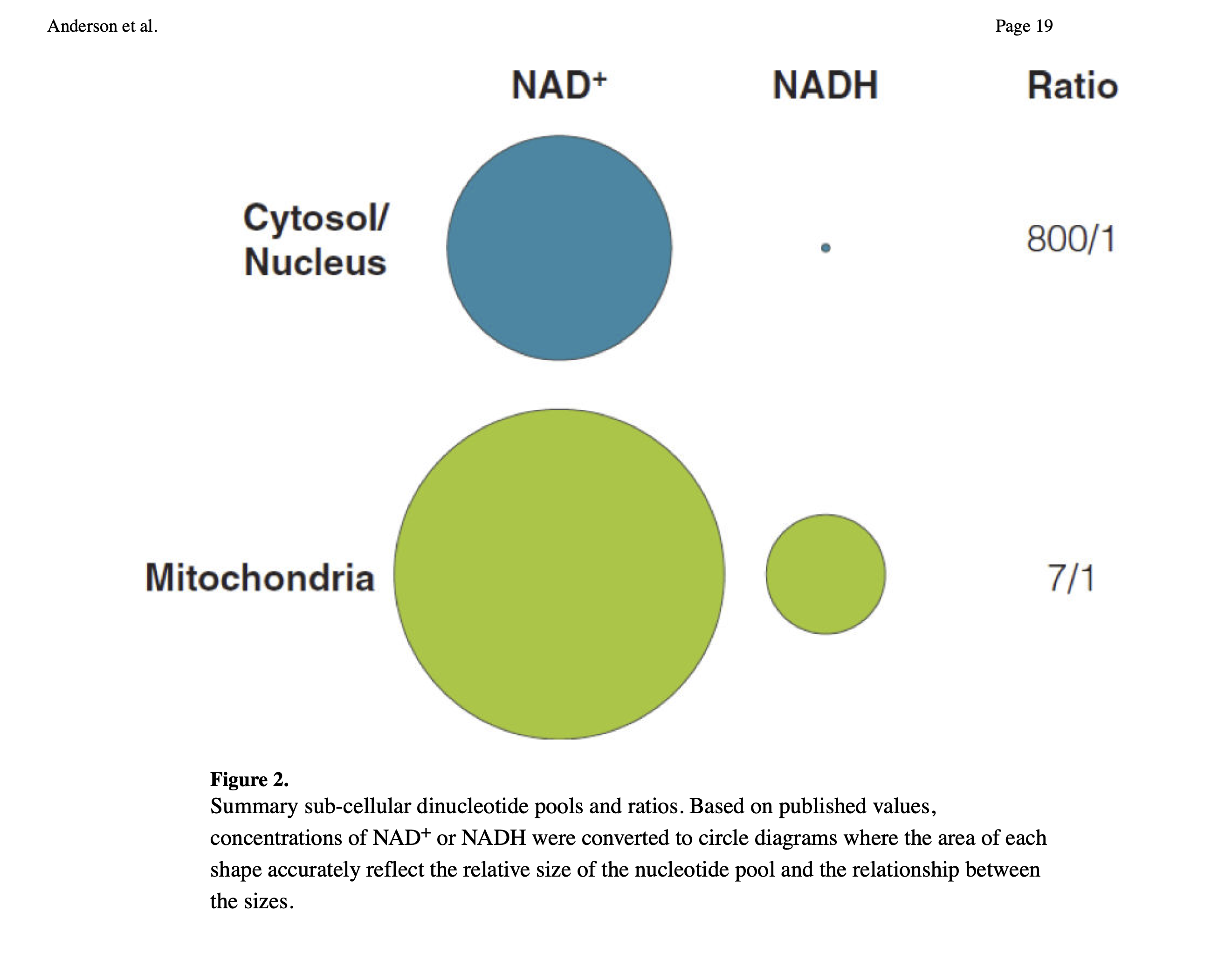
Since there is more NADH in mitochondria than in the cytosol and nuclear compartments, the more mitochondria a cell has the more global NADH there has to be. Maybe this means less NADH is used for converting AcAc into BOHB in those with more mitochondria (e.g. athletes). But I'm speculating.
Fat access at rest
A final idea to offer on the lower BOHB of LMHRs is that they simply have lower levels of FFAs (free fatty acids) in circulation as a function of fat mass. Compared to a fatter, but still metabolically healthy control, a leaner individual may have BOHB levels that depend more on their relative fat and protein intake, as fat mass is contributing less. In other words, a very lean individual could well be getting less energy from fat and more energy from protein than someone slightly fatter given the same dietary intake.
As to higher baseline glucose levels, seen in some (but not all!) LMHRs, it may simply be a combination of (1) lower basal insulin / higher hepatic insulin resistance, leading to less suppression of endogenous glucose production, (2) lower basal insulin / higher adipose insulin resistance, so that less is taken up into fat cells, and (3) higher fatty acid levels taken up into muscle and muscle mitochondria reciprocally downregulating glucose uptake there as well. That is, mildly elevated glucose levels (compared to the high carbohydrate condition) may simply be another concordant marker of "benevolent pseudo-diabetes" [Bla2019].
Further reading
Siobhan Huggins' primer on lipoproteins and energy transport
Footnotes
[1] The threshold amount of ketogenesis to be considered "in ketosis" is just a convention, although it's not completely arbitrary, but based on clinical effects. I follow prior researchers in using a blood BOHB level of 0.5 mM to mark the onset of ketosis ([Gue2003], [Gib2015]) but values as low as 0.2 have been used ([Mit1995]), and it's good to keep in mind that it's not really an on / off switch.
[2] Other measures of keto-adaptation
There are other measures that have been proposed for indicating keto-adaptation. One interesting one to me is uric acid.
Phinney has proposed the re-normalisation of uric acid (after the initial rise) as a marker of keto-adaptation, since excretion of uric acid and ketones is competitive in the kidneys. This takes up to several weeks.
I like this explanation in part because of my own theory that humans developed high uric acid in part to maintain ketosis [OHe2020].
Another one I've considered is respiratory quotient (RQ). RQ can tell you if you're primarily using fat for energy. RQ for AcAc and BOHB is 1.00 and 0.89 respectively [Fra1983], and this is already accounted for in the RQ of fat, as shown in this figure from Arch et al. [Arc2006].

Whether you metabolise the fat all the way in one place, or partially in the liver, and then the rest of the way after transport to the brain or the periphery, is not something separably capturable by RQ, which is a whole body integration. As Frayn points out, though the time differential in the latter case has an effect as concentrations rise and fall [Fra1983].
So while RQ itself can be a good measure of fat burning, but not of ketonemia, surprisingly, with the dimension of time it might give some clues to that as well.
[3] Also called Hindsight Bias
[4] Measuring acetone
Acetone can be measured in the breath. Since the Ketonix device first came out about a decade ago, many breath acetone (BrAce) metres have hit the market. How do they compare? There are several features that might matter to you when you are looking for a BrAce metre. The ones that matter most to me are accuracy and unit of measurement.
I do not want a device that doesn't offer ppm (parts per million), because that is the unit reported in scientific studies that I want to compare to. This rules out a couple of the currently popular devices that make their own proprietary scale, and just tell you "how ketogenic" they think you are based on equations they don't reveal anything about. Worse, the aim of these formulas seems to be to try to approximate corresponding BOHB. I already have a way to measure BOHB. I want a BrAce metre to measure acetone, not to approximate BOHB.
A second important property is accuracy. As far as I can tell, there is only one device on the market that addresses the problem of air flow. A device that merely accumulates a measurement over time has the problem that it would matter how long or hard you blew into the device, making it extremely difficult to tell the true value. Biosense is the only BrAce metre that addresses this problem, which it does by repeatedly sampling the air. (I have no affiliation.) This technology increases the price of the device, but I think it's worth it for accuracy.
A third important feature is range. I have used very inexpensive ppm devices sold as alcohol breathalysers. (Note, I've been told the new breathalysers no longer detect acetone, so this trick may no longer work.) These worked, except that I don't think they are accurate outside of the very narrow range people care about with alcohol: legal to drive or not. So I think it reflected more of a binary yes/no than a scale.
The Biosense, unfortunately, has a more limited range than I'd prefer. It is fine if you are in mild to moderate ketosis, but it maxes out fairly low, making it uninformative for making distinctions within the high levels of ketosis you would be in on a fast, or therapeutic levels of ketosis. They may address this is subsequent models.
I also have a Ketoscan device, which does give ppm and a decent range in the high levels, but given the air sampling problem, I don't necessarily trust its accuracy. It also needs to be sent in, maybe once a year, for "recalibration".
Finally note that the correspondence between plasma acetone and blood acetone is not perfectly linear although in the nutritional ketogenic range it may be close enough [Mus2006].
[5] Note that Dave Feldman recently replicated and extended the lathosterol results as reported in his labs from an experiment comparing hypocaloric and hypercaloric ketogenic diets. All the data is linked to from this post about the oxLDL results.
[6] I would love to find more data about specific levels and ratios at milder clinical ketosis. For example, what are the AcAc levels typically seen in those with BOHB between 0.5 and 1.5 mmol? I was unable to find data for this and would welcome some!
End-to-end citations
[Arc2006] Arch, J. R. S., D. Hislop, S. J. Y. Wang, and J. R. Speakman. “Some Mathematical and Technical Issues in the Measurement and Interpretation of Open-Circuit Indirect Calorimetry in Small Animals.” International Journal of Obesity 30, no. 9 (September 2006): 1322–31. https://doi.org/10.1038/sj.ijo.0803280.
[Bea1985] Beattie, M. A., and W. W. Winder. “Attenuation of Postexercise Ketosis in Fasted Endurance-Trained Rats.” American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 248, no. 1 (January 1, 1985): R63–67. https://doi.org/10.1152/ajpregu.1985.248.1.R63.
"Endurance-trained animals and human subjects have been reported to exhibit a lesser degree of postexercise ketosis than nontrained controls. We have studied the mechanism of this adaptation. Trained (2 h/day, 6 wk) and nontrained rats were fasted overnight and then run at 16 m/min up a 15% grade for 90 min. Trained rats had lower blood 3-hydroxybutyrate during exercise and during a 90-min postexercise period than nontrained rats. Liver malonyl coenzyme A (CoA), carnitine, and glycogen were not significantly different in the two groups at any time during and after exercise. Therefore these factors cannot be responsible for the difference in ketonemia. Plasma free-fatty acids and hepatic adenosine 3',5'-cyclic monophosphate were elevated in nontrained rats with respect to trained rats. These two differences could conceivably be responsible for a different ketogenic rate. In addition, 3-ketoacid CoA transferase activity of gastrocnemius muscle was increased by training. The increase in ketone oxidizing enzymes of muscle may also be partially responsible for the training-induced attenuation of postexercise ketonemia in these fasted rats."
[Ber1984] Bergstrom, J. D., G. A. Wong, P. A. Edwards, and J. Edmond. “The Regulation of Acetoacetyl-CoA Synthetase Activity by Modulators of Cholesterol Synthesis in Vivo and the Utilization of Acetoacetate for Cholesterogenesis.” The Journal of Biological Chemistry 259, no. 23 (December 10, 1984): 14548–53.
"This study shows that AcAc is utilized preferentially for cholesterol synthesis and that the regulation of AcAc-CoA synthetase is linked to the regulation of cholesterol synthesis. These studies suggest that the flux of AcAc-CoA production via AcAc-CoA synthetase may be influential in determining the rate of cholesterol synthesis. The importance of AcAc- CoA synthetase to cholesterol production revolves around the fundamental and as yet unanswered question of how impor- tant AcAc may be as a substrate for hepatic cholesterol synthesis. Since AcAc is principally derived from the hepatic oxidation of fatty acids and then can be utilized preferentially for cholesterol synthesis, it follows that AcAc may be an important intermediate in the conversion of fat to cholesterol. It should be kept in mind that the role of AcAc as a substrate for cholesterol synthesis may change in varying physiological situations."
[Bla2019] Blagosklonny, Mikhail V. “The Mystery of the Ketogenic Diet: Benevolent Pseudo-Diabetes.” Cell Cycle 18, no. 18 (September 17, 2019): 2157–63. https://doi.org/10.1080/15384101.2019.1644765.
[Bon1974] Boninsegna, A., G. Federspil, and C. De Palo. “The Effect of Muscular Exercise on Free Fatty Acids, Acetoacetate and 3-Hydroxybutyrate Blood Levels.” Hormone and Metabolic Research = Hormon- Und Stoffwechselforschung = Hormones Et Metabolisme 6, no. 6 (November 1974): 488–91. https://doi.org/10.1055/s-0028-1093809.
[Bro2010] Browning, Jeffrey D., and Jay D. Horton. “Fasting Reduces Plasma Proprotein Convertase, Subtilisin/Kexin Type 9 and Cholesterol Biosynthesis in Humans.” Journal of Lipid Research 51, no. 11 (November 2010): 3359–63. https://doi.org/10.1194/jlr.P009860.
"Proprotein convertase, subtilisin/kexin type 9 (PCSK9), a key regulator of plasma LDL-cholesterol (LDL-c) and cardiovascular risk, is produced in liver and secreted into plasma where it binds hepatic LDL receptors (LDLR), leading to their degradation. PCSK9 is transcriptionally activated by sterol response element-binding protein (SREBP)-2, a transcription factor that also activates all genes for cholesterol synthesis as well as the LDLR. Here we investigated the relationship between plasma PCSK9 levels and the lathosterol-to-cholesterol ratio, a marker of cholesterol biosynthesis, in 18 healthy subjects during a 48 h fast. In all individuals, plasma PCSK9 levels declined steadily during the fasting period, reaching a nadir at 36 h that was ~58% lower than levels measured in the fed state ( P < 0.001). Similarly, the lathosterol-to-cholesterol ratio declined in parallel with plasma PCSK9 concentrations during the fast, reaching a nadir at 36 h that was ~28% lower than that measured in the fed state ( P = 0.024).In summary, fasting has a marked effect on plasma PCSK9 concentrations, which is mirrored by measures of cholesterol synthesis in humans. Inasmuch as cholesterol synthesis and PCSK9 are both regulated by SREBP-2, these results suggest that plasma PCSK9 levels may serve as a surrogate marker of hepatic SREBP-2 activity in humans.—Browning, J. D., and J. D. Horton. Fasting reduces plasma proprotein convertase, subtilisin/kexin type 9, and cholesterol biosynthesis in humans."
[Cre2018] Creighton, Brent C, Parker Neil Hyde, Carl M Maresh, William J Kraemer, Stephen D Phinney, and Jeff S Volek. “Paradox of Hypercholesterolaemia in Highly Trained, Keto-Adapted Athletes.” BMJ Open Sport & Exercise Medicine 4, no. 1 (October 2018): e000429. https://doi.org/10.1136/bmjsem-2018-000429.
"Although ketone synthesis occurs exclusively in the mitochondria and cholesterol synthesis in the cytoplasm (37) it is possible for some acetoacetate generated during ketogenesis to diffuse out of the mitochondria and be converted to acetoacetyl-CoA in the cytosol via the action of acetoacetyl-CoA synthetase (38). Thus, the higher flux of fatty acids and ketogenesis in LC athletes in the context of overall high energy expenditures could contribute to increased endogenous synthesis of cholesterol by enhancing the cytosolic substrate pool. Increased circulating lathosterol and to a lesser extent desmosterol expressed relative to total cholesterol are markers of de novo cholesterol synthesis (39). Lathosterol was lower in LC athletes, indicating that cholesterol overproduction is likely not a major contributor to hypercholesterolaemia (40)."
[Cot2013] Cotter, David G., Rebecca C. Schugar, and Peter A. Crawford. “Ketone Body Metabolism and Cardiovascular Disease.” American Journal of Physiology-Heart and Circulatory Physiology 304, no. 8 (April 15, 2013): H1060–76. https://doi.org/10.1152/ajpheart.00646.2012.
[End1982] Endemann, G., P. G. Goetz, J. Edmond, and H. Brunengraber. “Lipogenesis from Ketone Bodies in the Isolated Perfused Rat Liver. Evidence for the Cytosolic Activation of Acetoacetate.” Journal of Biological Chemistry 257, no. 7 (April 10, 1982): 3434–40.
[Eva2017] Evans, Mark, Karl E. Cogan, and Brendan Egan. “Metabolism of Ketone Bodies during Exercise and Training: Physiological Basis for Exogenous Supplementation.” The Journal of Physiology 595, no. 9 (May 1, 2017): 2857–71. https://doi.org/10.1113/JP273185.
"[U]nlike CHO and fat, there is progressive attenuation of the oxidation of KBs with rising ketonaemia, and thus the mobilisation of KBs is not the factor limiting oxidation in skeletal muscle. This attenuation of exercise-stimulated MCR suggests either that above a threshold concentration the capacity for skeletal muscle to oxidise KBs becomes saturated, and/or that hyperketonaemia itself is a self-inhibitory factor (Balasse & Fery, 1989). Mechanistically, this is likely to be mediated either through the inhibition of OXCT by elevated AcAc, and/or via FFA-mediated inhibition of ketolysis (Robinson & Williamson, 1980). This regulation is critical in the starvation response because the capacity of the liver to produce KBs closely matches the requirements of the brain to utilise KBs as an energy source (Robinson & Williamson, 1980). Therefore, excessive oxidation by working muscle would threaten survival, whereas its inhibition spares circulating substrate for the brain (Hagenfeldt & Wahren, 1971; Fery & Balasse,1983)."
[Fra1983] Frayn, K. N. “Calculation of Substrate Oxidation Rates in Vivo from Gaseous Exchange.” Journal of Applied Physiology 55, no. 2 (August 1, 1983): 628–34. https://doi.org/10.1152/jappl.1983.55.2.628.
"When the ketone bodies previously stored are being utilized, then the effect will be to raise the RQ, the RQ for oxidation of AcAc being 1.00 and 3-OHB being 0.89 as shown in Eqs. 16 and 17
"AcAc (C4H503-) + H+ + 4 O2 -+ 4 CO2 + 3 H20 (16)
"2 3-OHB (C4H703-) + 2 H+ + 9 O2 -+ 8 CO2 + 8 H20 (17)"
[Gee1983] Geelen, M.J.H., M. Lopes-Cardozo, and J. Edmond. “Acetoacetate: A Major Substrate for the Synthesis of Cholesterol and Fatty Acids by Isolated Rat Hepatocytes.” FEBS Letters 163, no. 2 (November 14, 1983): 269–73. https://doi.org/10.1016/0014-5793(83)80833-3.
[emphasis mine]
"From a physiological viewpoint it seems unlikely that in one cell compartment, the mitochondrion, fatty acids are degraded to yield ketone bodies which in another cell compartment, the cytosol, are utilized for fatty acid synthesis. It is possible, however, that ketone bodies function as intermediates in the formation of long-chain fatty acids from fatty acids of short and medium-chain length. Preferential use of ketone bodies for hepatic cholesterogenesis is another possibility. Arguments in favour of the latter hypothesis are:
"(i) Acetoacetyl-CoA synthetase [5] and HMG-CoA reductase [7] have a similar diurnal rhythm, suggesting that acetoacetate can serve to carry Cd units from mitochondria to the cytosol where the acetoacetyl moiety can be incorporated directly into cytosolic HMG-CoA and then into cholesterol; (ii) Ketone-body utilization has been associated with cholesterogenesis in extra-hepatic tissues [8,9] and in isolated perfused rat liver [6]"
...
"Acetoacetate has been considered as a carrier for acetyl units in extra-hepatic tissues [8,20]. Our data, which corroborate the work in [6] with perfused liver, indicate that acetoacetate can fulfill a similar carrier function in rat liver. The preferential incorporation of acetoacetate into cholesterol over fatty acids observed both in brain [8,9] and in liver ([6], here) suggests that acetoacetate has a specific function in cholesterogenesis."
...
"In conclusion, our results confirm the observations in [6] that the liver can utilize ketone bodies for the synthesis of lipids, especially of cholesterol. Furthermore, our data extend their results: (i) Ketone-body production and utilization can proceed in the same cell type, i.e., the parenchymal liver cell; ii) The contribution of acetoacetate to lipid synthesis in hepatocytes is influenced by the contribution of other lipogenic substrates and by the physiological state of the donor animal; (iii) Ketone bodies are preferred to other lipogenic substrates for the de novo synthesis of cholesterol."
[Gib1986] Gibbons, G F, C P Attwell Thomas, and C R Pullinger. “The Metabolic Route by Which Oleate Is Converted into Cholesterol in Rat Hepatocytes.” Biochemical Journal 235, no. 1 (April 1, 1986): 19–24. https://doi.org/10.1042/bj2350019.
[Gib2015] Gibson, A. A., R. V. Seimon, C. M. Y. Lee, J. Ayre, J. Franklin, T. P. Markovic, I. D. Caterson, and A. Sainsbury. “Do Ketogenic Diets Really Suppress Appetite? A Systematic Review and Meta-Analysis: Do Ketogenic Diets Really Suppress Appetite?” Obesity Reviews 16, no. 1 (January 2015): 64–76. https://doi.org/10.1111/obr.12230.
[Gue2003] Guerci, B., M. Benichou, M. Floriot, P. Bohme, S. Fougnot, P. Franck, and P. Drouin. “Accuracy of an Electrochemical Sensor for Measuring Capillary Blood Ketones by Fingerstick Samples During Metabolic Deterioration After Continuous Subcutaneous Insulin Infusion Interruption in Type 1 Diabetic Patients.” Diabetes Care 26, no. 4 (April 1, 2003): 1137–41. https://doi.org/10.2337/diacare.26.4.1137.
[Hag1971] Hagenfeldt, L., and J. Wahren. “Human Forearm Muscle Metabolism during Exercise VI. Substrate Utilization in Prolonged Fasting.” Scandinavian Journal of Clinical and Laboratory Investigation 27, no. 4 (January 1971): 299–306. https://doi.org/10.3109/00365517109080222.
[Kal1999] Kalapos, M.P. “Possible Physiological Roles of Acetone Metabolism in Humans.” Medical Hypotheses 53, no. 3 (September 1999): 236–42. https://doi.org/10.1054/mehy.1998.0752.
"Summary: Recently, the metabolic routes for acetone metabolism have been described. Therefore, acetone cannot further be regarded as a waste product of metabolism. However, its physiological role in biochemical machinery is not clear. Here, an integrative model for the role of acetone metabolism is presented that orders the events occurring in acetonemia in sequence: (i) acetone participates in pH regulation; and (ii) acetone degradation in the liver both contributes to glucostatic function of the liver and provides C3 fragments to peripheral tissues as additional fuel. The model raises a novel approach to the study of the physiological role of acetone metabolism."
...
"The major part of ketogenesis takes place in the liver and the increased β-oxidation of fatty acids results in an increase in the concentration of NADH+H+, which probably through the malate-aspartate shuttle raises the redox potential in the cytosol, as well. In addition, since NADH+H+ is certain to be the direct hydrogen donor for acetoacetate, in the liver, consequently the disturbance of the NADH+H+/NAD+ ratio likely prompts the reduction of acetoacetate to β-hydroxybutyrate that is as an indicator of hepatic intramitocondrial redox-state, β-hydroxy butyrate/acetoacetate ratio is augmented. In the peripheral tissues, the reverse reaction is promoted, and in this way the hepatic and extrahepatic mitochondrial redox-status can be poised (25)." ...
"[W]hen ketone body production exceeds the degrading capacity, the accumulating acetoacetic acid presents a new challenge to the pH regulatory system. Acetone production and its further degradation to C3 fragments fulfill two purposes: the maintenance of pH buffering capacity and provision of fuel for peripheral tissues."
[Mit1995] Mitchell GA, Kassovska-Bratinova S, Boukaftane Y, et al. Medical aspects of ketone body metabolism. Clinical and Investigative medicine. Medecine Clinique et Experimentale. 1995 Jun;18(3):193-216.
Mus2002 Musa-Veloso, Kathy, Sergei S Likhodii, and Stephen C Cunnane. “Breath Acetone Is a Reliable Indicator of Ketosis in Adults Consuming Ketogenic Meals.” The American Journal of Clinical Nutrition 76, no. 1 (July 1, 2002): 65–70. https://doi.org/10.1093/ajcn/76.1.65.
"A dynamic equilibrium exists between plasma β-hydroxybutyrate and plasma acetoacetate. Plasma acetoacetate and β-hydroxybutyrate both increase acutely after the consumption of a ketogenic meal. Between meals, plasma acetoacetate but not β-hydroxybutyrate is directly metabolized for energy and, at other times, may also be used for fatty acid synthesis. Plasma β-hydroxybutyrate has been referred to as a “dead-end metabolite” in that its only known role is interconversion with acetoacetate(28). Thus, β-hydroxybutyrate itself does not provide energy unless it is first converted to acetoacetate via β-hydroxybutyrate dehydrogenase (28). In a sense, plasma β-hydroxybutyrate can be thought of as a ketone reserve; when plasma acetoacetate concentrations begin to decrease, more of it is produced from β-hydroxybutyrate. The equilibrium between plasma acetoacetate and plasma β-hydroxybutyrate was apparent neither in breath acetone nor in urinary acetoacetate."
[Mus2006] Musa-Veloso, Kathy, Sergei S. Likhodii, Exequiel Rarama, Stephanie Benoit, Yeou-mei Christiana Liu, Dominic Chartrand, Rosalind Curtis, et al. “Breath Acetone Predicts Plasma Ketone Bodies in Children with Epilepsy on a Ketogenic Diet.” Nutrition 22, no. 1 (January 2006): 1–8. https://doi.org/10.1016/j.nut.2005.04.008.
"Other studies have shown a positive linear relation be-tween breath acetone up to 25 000 nmol/L and plasmaacetone up to 15 mmol/L[18–21]. In the present study,plasma acetone up to 9 mmol/L was positively but curvi-linearly related to breath acetone up to 18 000 nmol/L"
[Nea2009] Neal, Elizabeth G., Hannah Chaffe, Ruby H. Schwartz, Margaret S. Lawson, Nicole Edwards, Georgiana Fitzsimmons, Andrea Whitney, and J. Helen Cross. “A Randomized Trial of Classical and Medium-Chain Triglyceride Ketogenic Diets in the Treatment of Childhood Epilepsy.” Epilepsia 50, no. 5 (2009): 1109–17. https://doi.org/10.1111/j.1528-1167.2008.01870.x.
[OHe2018] O’Hearn, L. Amber. “Evidence on Chronic Ketosis in Traditional Arctic Populations.” Journal of Evolution and Health 3, no. 1 (February 19, 2019). https://doi.org/10.15310/2334-3591.1101.
[OHe2020] O’Hearn, L Amber. “My Case Against Uricase: A Critical Examination of Hypotheses.” Journal of Evolution and Health: An Ancestral Health Society Publication 4, no. 1 (February 4, 2020). https://doi.org/10.15310/J34145990.
[Owe2005] Owen, Oliver E. “Ketone Bodies as a Fuel for the Brain during Starvation.” Biochemistry and Molecular Biology Education 33, no. 4 (2005): 246–51. https://doi.org/10.1002/bmb.2005.49403304246.
[Puc2017] Puchalska, Patrycja, and Peter A. Crawford. “Multi-Dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics.” Cell Metabolism 25, no. 2 (February 7, 2017): 262–84. https://doi.org/10.1016/j.cmet.2016.12.022.
"While the physiological significance is yet to be established, ketones can serve as anabolic substrates even in the liver. In artificial experimental contexts, AcAc can contribute to as much as half of newly synthesized lipid, and up to 75% of new synthesized cholesterol (Endemann et al., 1982; Geelen et al., 1983; Freed et al., 1988). Because AcAc is derived from incomplete hepatic fat oxidation, the ability of AcAc to contribute to lipogenesis in vivo would imply hepatic futile cycling, where fat-derived ketones can be utilized for lipid production, a notion whose physiological significance requires experimental validation, but could serve adaptive or maladaptive roles (Solinas et al., 2015). AcAc avidly supplies cholesterogenesis, with a low AACS Km-AcAc (~50 µM) favoring AcAc activation even in the fed state (Bergstrom et al., 1984). The dynamic role of cytoplasmic ketone metabolism has been suggested in primary mouse embryonic neurons and in 3T3-L1 derived-adipocytes, as AACS knockdown impaired differentiation of each cell type (Hasegawa et al., 2012a; Hasegawa et al., 2012b). Knockdown of AACS in mice in vivo decreased serum cholesterol (Hasegawa et al., 2012c)."
[Rei1979] Reichard, G. A., A. C. Haff, C. L. Skutches, P. Paul, C. P. Holroyde, and O. E. Owen. “Plasma Acetone Metabolism in the Fasting Human.” Journal of Clinical Investigation 63, no. 4 (April 1, 1979): 619–26. https://doi.org/10.1172/JCI109344.
"The appearance of radioactivity in plasma glucose, lipids, and proteins supports the results of somewhat similar studies in animals in which labeled carbon of acetone was found in cholesterol (25), liver glycogen (26, 28), and various amino acids derived from liver and carcass protein (26,28). Incorporation of radioactivity fromacetone into glucose suggests the possibility of gluconeogenesis from the compound, or a metabolite(s) of the compound, a process for which evidence has been obtained in rats(28) and ketotic cows (29,30). On the basis of our specific activity data, we have calculated that ≊4-11% of plasma glucose production could theoretically be derived from acetone. The value of 11% was obtained in 21-d fasted obese subjects in whom net splanchnic and renal glucose release rates of ≊370 μmol/min, requiring 80 μmol/min glucose equivalents of acetone or 59% of the plasma acetone production rates measured in these subjects. It is recognized that these calculations are based on an unproven assumption that net glucose synthesis from acetone can occur in the human. They were done, however, to gain some insight into the possible importance of this pathway as a mechanism for acetone disposal."
[Sal1989] Salam, W H, H G Wilcox, L M Cagen, and M Heimberg. “Stimulation of Hepatic Cholesterol Biosynthesis by Fatty Acids. Effects of Oleate on Cytoplasmic Acetoacetyl-CoA Thiolase, Acetoacetyl-CoA Synthetase and Hydroxymethylglutaryl-CoA Synthase.” Biochemical Journal 258, no. 2 (March 1, 1989): 563–68. https://doi.org/10.1042/bj2580563.
"The effects of oleic acid on the activities of cytosolic HMG-CoA (3-hydroxy-3-methylglutaryl-CoA) synthase, AcAc-CoA (acetoacetyl-CoA) thiolase and AcAc-CoA synthetase, as well as microsomal HMG- CoA reductase, all enzymes in the pathway of cholesterol biosynthesis, were studied in the isolated perfused rat liver. Oleic acid bound to bovine serum albumin, or albumin alone, was infused for 4h at a rate sufficient to sustain an average concentration of 0.61+0.05mm fatty acid during the perfusion. Hepatic cytosol and microsomal fractions were isolated at the termination of the perfusion. Oleic acid simultaneously increased the activities of the cytosolic cholesterol-biosynthetic enzymes 1.4-2.7-fold in livers from normal fed rats and from animals fasted for 24 h. These effects were accompanied by increased net secretion by the liver of cholesterol and triacylglycerol in the very-low-density lipoprotein(VLDL). We confirmed the observations reported previously from this laboratory of the stimulation by oleic acid of microsomal HMG-CoA reductase. In cytosols from perfused livers, the increase in AcAc-CoA thiolase activity was characterized by an increase in Vmax without any change in the apparent Km of the enzyme for AcAc-CoA. In contrast, oleic acid decreased the Km of HMG-CoA synthase for Ac-CoA, without alteration of the Vmax of the enzyme. The Vmax of AcAc-CoA synthetase was increased by oleic acid, and there was a trend towards a small increase in the Km of the enzyme for acetoacetate. These data allow us to conclude that the enzymes that supply the HMG-CoA required for hepatic cholesterogenesis are stimulated, as is HMG-CoA reductase, by a physiological substrate, fatty acid, that increases rates of hepatic cholesterol synthesis and cholesterol secretion. Furthermore, we suggest that these effects of fatty acid on hepatic cholesterol metabolism result from stimulation of secretion of triacylglycerol in the VLDL by fatty acids, and the absolute requirement of cholesterol as an important structural surface component of the VLDL necessary for transport of triacylglycerol from the liver."
[...]
"Oleate was reported in a previous study to be more effective than either palmitate or linoleate in stimulating hepatic HMG-CoA reductase activity [2,4], although the molar ratio of hepatic output of cholesterol/triacylglycerol was in the order palmitate > oleate > linoleate."
[Vol2011] Volek, Jeff, Phd Stephen D. Phinney MD, Rd Jeff S. Volek Phd, and Stephen D. Phinney. The Art and Science of Low Carbohydrate Living: An Expert Guide to Making the Life-Saving Benefits of Carbohydrate Restriction Sustainable and Enjoyable. Beyond Obesity, 2011.
"Beta-hydroxybutyrate and acetoacetate are made in the liver in about equal proportions, and both are initially promptly oxidized by muscle. But over a matter of weeks, the muscles stop using these ketones for fuel. Instead, muscles take up acetoacetate, reduce it to beta-hydroxybutyrate, and return it back into the circulation. Thus after a few weeks, the predominant form in the circulation is beta-hydroxybutyrate, which also happens to be the ketone preferred by brain cells... The result of this process of keto-adaptation is an elegantly choreographed shuttle of fuel from fat cells to liver to muscle to brain."
[Vol2016] Volek, Jeff S., Daniel J. Freidenreich, Catherine Saenz, Laura J. Kunces, Brent C. Creighton, Jenna M. Bartley, Patrick M. Davitt, et al. “Metabolic Characteristics of Keto-Adapted Ultra-Endurance Runners.” Metabolism 65, no. 3 (March 1, 2016): 100–110. https://doi.org/10.1016/j.metabol.2015.10.028.
[Whi2011] White, Hayden, and Balasubramanian Venkatesh. “Clinical Review: Ketones and Brain Injury.” Critical Care 15, no. 2 (2011): 219. https://doi.org/10.1186/cc10020.
"AcAc is the central ketone body. AcAc is reduced to BHB by β-hydroxybutyrate dehydrogenase (BHD) in a NADH-requiring reversible reaction [3]:
"(R)-3-Hydroxybutyrate + NAD⁺ ⟷ acetoacetate + NADH + H⁺
"The extent of this reaction depends on the state of the cellular pool of NAD+ [4]. When highly reduced, most or all ketones will be in the form of BHB. The ratio of BHB to AcAc reflects the redox state within the mitochondrial matrix [5]. BHB is nonvolatile and is chemically stable. This ketone body's only metabolic fate is interconversion with AcAc. Once synthesized, ketones diffuse into the circulation."
[Whi2012] White, Amanda T., and Simon Schenk. “NAD+/NADH and Skeletal Muscle Mitochondrial Adaptations to Exercise.” American Journal of Physiology-Endocrinology and Metabolism 303, no. 3 (March 27, 2012): E308–21. https://doi.org/10.1152/ajpendo.00054.2012.